
Abstract
In this study, fibrin, which is superior to fibrinogen in both structural and functional properties, has for the first time been electrospun successfully into uniform nano fibers resembling the extracellular matrix (ECM). The methods of fabrication and characterization of this unique scaffold are presented. Using poly (vinyl) alcohol as an “electrospinning-driving” polymer, we have developed a novel method for the fabrication of fibrin into a nanofibrous scaffold for various tissue-engineering applications starting from human-plasma-derived fibrinogen and thrombin and combining these ingredients within the syringe of an electrospinning setup under high voltage. In this fashion, fibrin nanofibrous scaffold is produced in a one-step approach without the need for subsequent cross-linking by synthetic agents that compromise the biological properties of the scaffold. Characterization of the electrospun membrane was done by scanning electron microscopy (SEM), Fourier transform infrared spectroscopy, and fibrin specific phosphotungstic acid hematoxylin staining. SEM data revealed the formation of bead-free fibers with a dimension ranging from 50–500 nm, which exactly mimics the fiber diameter of native ECM. Cell attachment and proliferation studies revealed that the scaffold supports the attachment, spreading, and proliferation of human umbilical cord blood-derived mesenchymal stem cells.
Introduction
In the context of tissue engineering, biodegradable polymers offer a good choice for the fabrication of functional tissue constructs. Naturally derived polymers such as fibrin, collagen, and so on have the potential advantages of cell-specific interactions and hydrophilicity. Fibrin is a polypeptide consisting of the plasma components fibrinogen and thrombin. The unique polymerization mechanism of fibrin, permitting control over gelation time and network architecture by varying the reaction conditions, allows formation of soft substrates under physiological conditions. 8 In vivo, fibrin is formed by the thrombin mediated cleavage of fibrinopeptide A and B from the Aα and Bβ chains of fibrinogen, respectively. This causes subsequent conformational changes that generate fibrin monomer which has a great propensity to self-associate and form insoluble fibrin. The plasma transglutaminase, factor XIII, further cross links the fibrin monomer to form a stable clot. Fibrin as well as fibrinogen have vital functions in blood clotting, cellular interactions, inflammation, wound healing, angiogenesis, and so on.9,10 Moreover, fibrin is less immunogenic among the same species. The safety of fibrin for biomedical applications has been proved by its widespread use as hemostatic agents and tissue sealants in various clinical applications including cardiac, liver, and neuro surgery. Currently, the Food and Drug Administration (FDA) approved allogenic fibrin sealants such as Tisseel®, Crosseal® etc. are used in the clinics as hemostatic agents. In the last decade, fibrin has been suggested for use in a variety of tissue-engineered implants for cardiovascular, liver, skin, bone, and cartilage.11–16 Due to its angiogenic potential, fibrin is used as a coating material for vascular constructs to enhance endothelialization and homing of vascular progenitor cells on the synthetic surfaces.17,18 However, the shrinkage of fibrin gel on sheet formation as well as its rapid degradation before functional tissue formation can be the major impediments in the use of fibrin for preparing tissue-engineered scaffolds.12,13
To overcome some of these structural deficiencies of conventional fibrin coatings or sheets, we have, in this work, shown for the first time that fibrin nanofibrous structures mimicking the ECM can be fabricated by electro-spinning, a well-established technique for scalable nanofibrous structure processing.19,20 Such a nanofibrous scaffold structure prepared by electrospinning fibrin with other biodegradable polymers would change dynamically with time on polymer degradation, thereby allowing the seeded cells to proliferate and create their own natural ECM. 21 Though fibrinogen and collagen have been successfully electrospun22–24 for tissue-engineering applications, fibrinogen has poor mechanical properties, and attempts to crosslink fibrinogen with synthetic crosslinking agents after electrospinning have resulted in a compromise of the bioactivity of this scaffold. For example, scaffolds crosslinked with gluteraldehyde, genipin, and 1-ethyl-3-[3-dimethyl aminopropyl] carbodiimide hydrochloride resulted in incomplete scaffold remodeling with very little collagen deposition after 21 days of in vitro fibroblast culture. 25 In this study, we have adopted a technique of direct in situ electrospinning of fibrin using a biomimetic approach of combining fibrinogen (derived from natural human plasma that contains the clot stabilizing factor plasma transglutaminase, Factor XIII) with thrombin, within the electrospinning syringe under applied voltage. Fibrinogen taken in the syringe is in combination with a suitable carrier (namely poly (vinyl) alcohol [PVA], an FDA approved polymer) to aid in its electrospinning. We demonstrate the process, stability, and characterization of the biological properties of the ECM-like fibrin nanofibrous scaffolds.
Recent advancements in tissue engineering and regenerative medicine have highlighted human umbilical cord blood-derived mesenchymal stem cells (UCB-hMSCs) as a potential cell source owing to their capacity to differentiate into various tissue types such as bone, cartilage, adipose tissue, liver, neuron, and cardiac muscle.26,27 Compared with hMSCs derived from other cell sources, UCB-hMSCs have high availability, are ontogenically primitive, have less immunogenicity, can be isolated without risk to the donor, and have a high degree of expansion potential in comparison to other cell sources. 28 Hence, in this study, nanofibrous fibrin scaffolds seeded with UCB-hMSCs were examined for cell attachment, spreading, proliferation, and metabolic activity.
Materials and Methods
Materials
Fibrinogen was prepared from pooled fresh frozen plasma of healthy human volunteers by cryoprecipitation method. 29 Bovine thrombin was purchased from Merck. PVA (MW 50,000, degree of hydrolysis-85%–89%), and Calcium chloride (CaCl2) was obtained from Qualigens, Mumbai, India. Tetra methyl rhodamine isothiocyanate (TRITC)-Phalloidin, mouse anti-human vinculin, and 4′,6-diamidino-2-phenylindole (DAPI) were obtained from Sigma Aldrich. Alamar blue, Vybrant apoptosis assay kit, Iscove's modified Dulbecco's medium (IMDM), MSC specific fetal bovine serum (MSC-FBS), and antibiotics were from GIBCO, Invitrogen. Antibodies such as CD44-fluorescein isothiocyanate (FITC), CD73-phycoerythrin (PE), CD90-allophycocyanin (APC), CD29-PE, CD34-FITC, CD31-FITC, CD33-APC, and CD45-PE used for the characterization of UCB-hMSCs had been obtained from BD Biosciences.
Electrospinning process
Fibrinogen at different concentrations (25–50 mg/mL) was prepared in sterile distilled water. Thrombin (50–100 U/mL) was prepared in 35 mM CaCl2 solution. The solution viscosities were separately measured using DV II+ Pro digital viscometer. These two solutions were separately taken in two 2 mL syringes and connected to a delivery device having a common needle at the end of the syringes. This delivery device was positioned in a KD scientific syringe pump to dispense the solutions at a predetermined flow rate. The flow rate was adjusted in such a way that the fibrin formation takes place immediately before the formation of fiber jet. A high voltage power supply (Gamma high voltage) was used to apply a voltage of 15–30 kV to the 18 gauge blunt end needle fixed to the Y shaped connector attached with the syringes containing the solutions. In the next trial, PVA was used as an “electrospinning driving polymer.” About 12% PVA solution was prepared in sterile water at 60°C with continuous stirring for at least 2 h and then cooled to room temperature. Fibrinogen and thrombin in PVA solution were separately prepared and taken in two syringes. Electrospinning was conducted in a similar manner as just described. The concentration of PVA was varied from 4% to 8% to obtain uniform fibers with nano-scale dimensions. The diagrammatic representation of the electrospinning set up used is shown in Figure 1. Solutions were electrospun on to the aluminium foil attached to the grounded mandrel placed at 10–15 cm from the needle tip. The scaffolds were removed from the mandrel soon after electrospinning and stored in the refrigerator for future use.

Diagrammatic representation of the electrospinning process. Two syringes were connected to a common needle for the simultaneous delivery of the solutions of fibrinogen and thrombin in PVA, and a high-voltage power supply for charging the solution to 15–30 kV. PVA, poly (vinyl) alcohol.
Removal of PVA from PVA-fibrin scaffolds
To remove PVA, the electrospun mats were immersed in phosphate-buffered saline (PBS) under gentle shaking at 50 rpm in an incubator shaker for 24 h. After 24 h, the scaffolds were freeze dried and kept in the refrigerator for future use. The dry weight of the scaffold before and after removal of PVA was taken, and the percentage weight loss was calculated to ensure complete removal of PVA.
Structural and morphological characterization
Scanning electron microscopy
Morphological characterization of electrospun scaffolds was done using scanning electron microscopy (SEM, JSM-6490 LA; JEOL). The scaffolds were fixed on to an aluminium stub, sputter coated with platinum using JEOL (model JFC-1600) auto fine coater, and observed at an accelerating voltage of 15 kV.
Fourier transform infrared spectroscopy
FTIR spectra of PVA, Fibrin, and electrospun PVA-Fibrin scaffold were recorded between 4000 and 400 cm−1 with a resolution of 4 cm−1 and 100 scans per sample using a Perkin Elmer Spectrum RX1 apparatus.
Phosphotungstic acid hematoxylin staining
Mallory's phosphotungstic acid hematoxylin (PTAH) staining technique was used to confirm the presence of fibrin in the electrospun scaffolds. Briefly, the electrospun mats were fixed on cover slips and soaked in PBS by gentle agitation overnight to remove PVA. The cover slips were then stained with PTAH and observed under a light microscope.
Cell behavior on electrospun scaffolds
The concentrations of fibrinogen, thrombin, and PVA used to produce electrospun scaffolds for cell culture experiments were 50 mg/mL, 100 U/mL, and 60 mg/mL, respectively. The processing parameters used for electrospinning were 0.4 mL/h flow rate, 10 cm tip target distance, and 15 kV voltage. The scaffolds were subjected to ethylene oxide sterilization before cell seeding. Cell studies were carried out using UCB-hMSCs. Cells after the third passage were used for all experiments.
Isolation, culture, and characterization of UCB-hMSCs
Umbilical cord blood samples were obtained from healthy babies, born in AIMS hospital, with previous approval from Institutional ethical committee. Mononuclear cells from umbilical cord blood were isolated by Ficoll-Paque (Amersham-Pharmacia) density gradient centrifugation (1.077 g/cm3), and seeded on to noncoated tissue culture flasks (Becton Dickinson) in MSC specific culture medium. Nonadherent cells were removed by medium changes after allowing the cells to attach overnight. Thereafter, media changes were carried out twice in a week. MSC specific medium consists of IMDM and 20% MSC-FBS supplemented with 100 U/mL penicillin and 100 U/mL streptomycin. MSC phenotype was confirmed by the positive expression of surface markers such as CD73, CD90, CD44, and CD29 and negative expression of markers such as CD33, CD45, CD31, and CD34 by flow cytometry (FACS Aria II, BD Biosciences). Data analysis was done using FACS Diva software version 6.1 (BD Biosciences).
Cell attachment and spreading
Electrospun scaffolds were cut into 2 cm2 small pieces, and UCB-hMSCs were seeded on to the scaffolds at a seeding density of 25,000-cells/cm2. The cell-fiber constructs were then cultured in the MSC specific medium in a humidified incubator at 37°C with 5% CO2. For SEM analysis, the medium was removed after the required incubation period (6 and 48 h), and the scaffolds were washed with PBS before fixing in 2% buffered gluteraldehyde for 1 h at room temperature. After fixing, samples were washed with PBS and dehydrated with sequential rinses for 15 min each with 30%, 50%, 70%, 90%, and 100% ethanol. Samples were then imaged using SEM after sputter coating at an accelerating voltage of 8 kV.
For the analysis of cytoskeletal organization and focal adhesion, cells were stained using TRITC-Phalloidin for F-actin and mouse monoclonal anti-human vinculin, respectively, after 24 h of cell seeding. Scaffolds were gently washed with PBS, fixed in 4% paraformaldehyde in PBS for 15 min, and then extensively washed in PBS. Cells were then permeabilized with 0.5% Triton X-100 in PBS for 5 min, and washed again. Then, for F-actin, staining was done with a 50 μg/mL TRITC-conjugated phalloidin solution in PBS for 40 min at room temperature, and then extensively washed with PBS to remove unbound phalloidin conjugates. Nuclei were visualized after counter staining with DAPI. For vinculin staining, cells were treated with mouse monoclonal anti-human vinculin antibody (1:500 dilution) for 60 min at ambient temperature followed by FITC-conjugated anti-mouse IgG antibody (Sigma) for 30 min. Samples were then washed, dried, and mounted for imaging using a fluorescence microscope (Olympus BX 51) or confocal microscope (Leica TCS SP5 II).
Cell proliferation
Cell proliferation studies were carried out using DAPI staining and Alamar blue assay. For DAPI staining, 25,000 cells/cm2 were seeded on the scaffolds and cultured for 6, 48, and 96 h. After the culture period, cells were stained with DAPI (10 μg/mL) for 5 min. Samples were mounted after washing in PBS, and the nuclei were observed under a fluorescence microscope.
A nonradioactive cell proliferation assay based on Alamar blue was employed for the quantification of cell proliferation. About 25,000 cells/cm2 cells were seeded on the scaffolds and cultured for three time points (6, 48, and 96 h). After the required incubation period, the medium was removed, Alamar blue in phenol-red-free medium was added and incubated at the same culture conditions for 4 h. After incubation, 200 μL of the pink-colored reaction product from each cell-scaffold containing culture plate were transferred into a 96-well plate, and the absorbance was quantified using a UV-Vis spectrophotometer (Power wave XS, Biotek) at 570 and 600 nm. A standard curve was plotted with known number of cells treated in a similar way, and the number of cells after each incubation period was quantified from the standard curve.
Statistics
Student's t-test was used to perform the statistical analysis of the data. Data are expressed as mean±standard deviation, and p<0.01 is considered significant and indicated as *; p<0.001 is considered as of a higher significance and indicated as ** (n=4).
Results and Discussion
Optimization of fibrin electrospinning
Several parameters such as viscosity flow rate, electric field, and so on affect the electrospinning process. Viscosity of fibrinogen solution at different concentrations is given in Table 1. Viscosity of thrombin solution at all concentrations was seemed to be approximately 1cP, which is the same as the viscosity of water. Lower viscosity of the samples may be one of the contributing parameters for spraying and bead formation.
We assumed that the gelation of fibrin at the needle tip would increase the viscosity, which will aid in the spinning of fibrin. However, since thrombin's function in the formation of fibrin from fibrinogen is essentially enzymatic, even a minute amount of thrombin resulted in faster clot formation. Fibrin formation, in the phase beyond the “latent period,” is basically “first order.” 30 Immediate fibrin formation occurred at the needle tip, which made the spinning of fibrinogen and thrombin practically impossible at all concentrations. This condition caused spraying rather than spinning (Fig. 2).

Scanning electron micrograph of electrospun fibrin structure at control parameters (15–30 kV, 0.3–1.0 mL/h flow rate, 10–15 cm working distance, and 25–50 mg/mL fibrinogen solution and 50–100 U/mL thrombin solution) at different magnifications
Electrospinning of natural polymers including fibrinogen often requires highly volatile solvents such as 1, 1, 1, 3, 3, 3 Hexafluoro-iso propanol (HFIP). However, HFIP and other harsh organic solvents are toxic and may affect the biological activity of the natural fibrin scaffold. 31 Optimizing the finest conditions for a specific polymer solvent system is crucial to get predictable and functional tissue-engineering scaffolds. The consequence of adding a natural polymer component such as chitosan to PVA solution was previously experimented, 32 and an optimal concentration of PVA (8% wt/v in water) was used to obtain beadless fibers. Thus, we selected PVA as an “electrospinning driving” polymer in our study due to its simplicity of use and nontoxicity. Also, it has been suggested that hydroxyl compounds such as PVA reduce the attractive forces between fibrinogen molecules and diminish their tendency to form side-by-side association during clotting, thereby resulting in a fine network rather than coarse strands. 33 Another benefit of using PVA as a binder is that the presence of hydroxyl compound can also slightly delay the formation of fibrin so that we can adjust the flow rate in such a way that fibrin is formed just before the formation of fiber jet at the needle tip. We started the electrospinning with 8% PVA concentration. However, the addition of 8% PVA solution resulted in increased viscosity of PVA fibrinogen mixture, which caused resistance to the jet formation. This instability in jet formation resulted in both spinning and spraying. The morphology of the fiber mat obtained at this condition is shown in Figure 3a. The solidification of fiber jet as they got ejected from the needle tip resulted in patch-like appearance with beads in the fibrous mat.

Scanning electron micrograph of electrospun PVA-fibrin structure at
Electrospinning at a PVA concentration of 4% resulted in defects in either the form of beads or droplets (Fig. 3b). The lower concentration is a characteristic of electrospraying rather than electrospinning. 34 Since the PVA concentration was increased to 6%, neither droplets nor beads were observed and resulted in the formation of nanofibers with interconnected pores (Fig. 3c). The spinning parameters were found to be 15 kV voltage, 0.4 mL/h flow rate, and 10 cm tip target distance. The fiber diameter appeared in the range of 50–500 nm (Fig. 3d). This range has been shown to favor cell matrix interactions.35,36
Removal of PVA from PVA-Fibrin scaffold
For long-term stability of electrospun fibrinogen, scaffolds both in vivo and in vitro it needs to be cross linked. The common cross-linking agents used for fibrinogen scaffolds not only affect the integrity of the proteins and cells but also these are often toxic and should be removed or extracted before tissue engineering applications. Since fibrin is insoluble in water, there is no need to cross link fibrin. Also, the fibrinogen rich cryoprecipitate used in this study contain a considerable amount of plasma transglutaminase (Factor XIII), which is a natural cross-linking agent and clot-stabilizing factor. PVA, which is a water soluble polymer, was removed from the scaffold by immersing it in PBS. The percentage weight loss after removal of PVA was found to be 61%±1.4% (n=4), which is approximately equal to the percentage of PVA (55%) in the scaffold. The splitting off of fibrinopeptide A and fibrinopeptide B from the fibrinogen molecule during fibrin formation may be the reason for the slight increase in actual percentage of weight loss during removal of PVA.
SEM images reveal that fibrin is retained in the scaffold and preserves the nanofibrous structure (Fig. 4b). The presence of fibrin in the scaffold is further confirmed by Fourier transform infrared spectroscopy (FTIR) and fibrin specific staining.

Scanning electron micrograph of electrospun PVA-fibrin structure at 6% PVA concentration
Chemical composition of electrospun scaffold
FTIR spectroscopy
The formation of fibrin in the mixed composition of fibrin containing scaffold was verified by ATR-FTIR spectrometry. Figure 5 shows the IR spectra of pure PVA, pure fibrin, and the PVA-Fibrin composite. The spectra exhibit characteristic bands that primarily result from vibrations in the peptide linkages. Spectra of composite membrane revealed overlapping peaks characteristic of fibrin at wave numbers of 3310 cm−1 (N-H stretch), 1649 cm−1 (amide I band), and 1500 cm−1 (amide II band), thus verifying that the composite scaffold is a mixture of fibrin and PVA. The bands at 1460 and 1390 cm−1 are assigned to the CH2 deformation and to vibrations of the aminoacid side chains, respectively. The amide III band is located at 1250 cm−1. 37
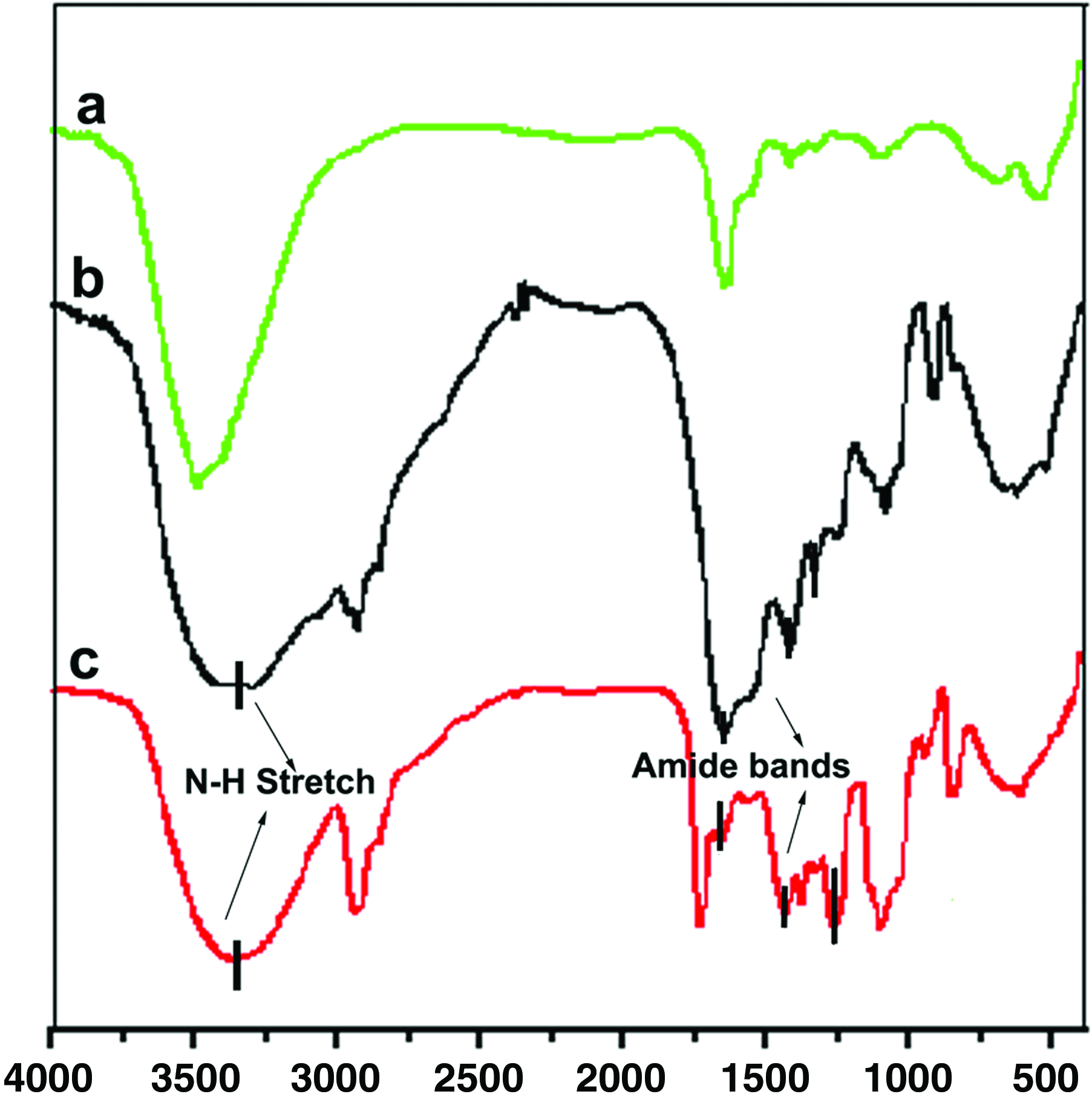
FTIR spectra of
PTAH staining
PTAH staining is a typical demonstration technique of fibrin. PTAH is a polychrome stain that gives blue color to fibrin. Figure 6a is the PTAH stained PVA-fibrin scaffold before the removal of PVA. Since PVA is highly hydrophilic, it absorbs water and swells up on absorbing water. The presence of fibrin can be confirmed by the characteristic blue color but, the nanofibers are not clearly distinct. When PVA is removed from the scaffold, the fibrin fibers are more clearly seen (Fig. 6b).
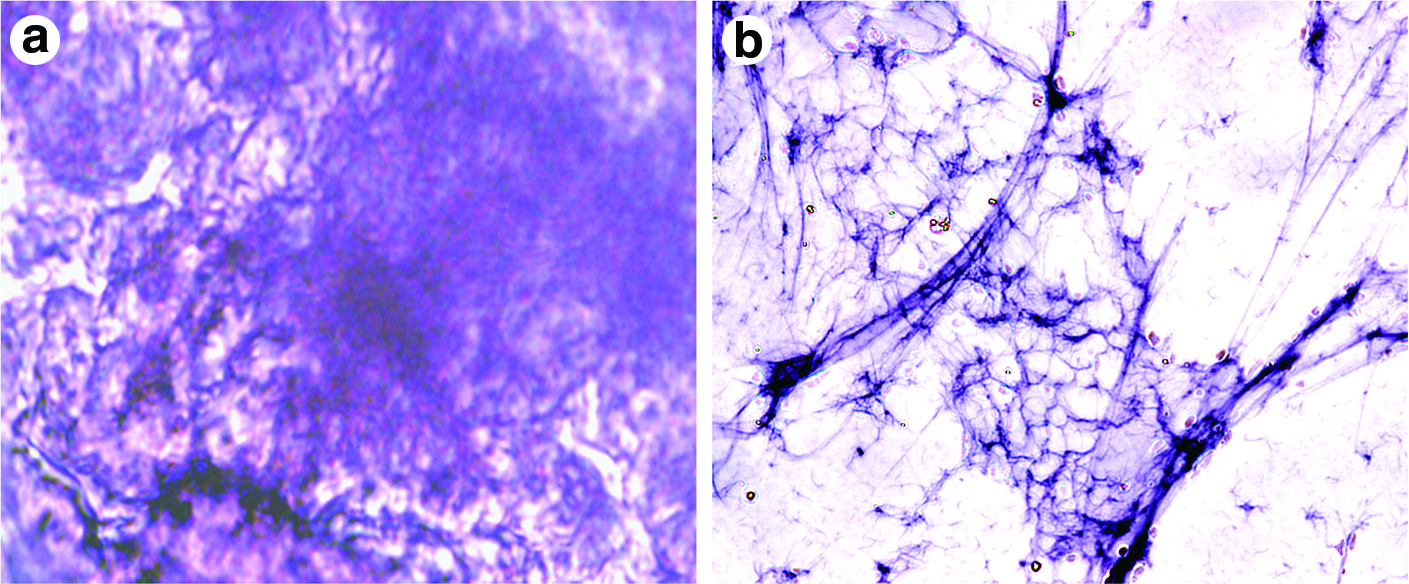
Light microscopic image of
Cell behavior on electrospun scaffolds
The capabilities of UCB-hMSCs to self-renew and differentiate into multiple lineages of tissues of mesenchymal origin draw a lot of scientific attention in the context of tissue-engineering and cell-based therapies. Since umbilical cord blood is also a rich source of hematopoietic stem cells, the phenotype of MSCs were confirmed by the positive expression of surface markers such as CD44, CD90, CD29, and and CD73 and the negative expression of CD31, CD34, CD33, and CD45 by flow cytometric analysis (Fig. 7; Supplementary data, available online at
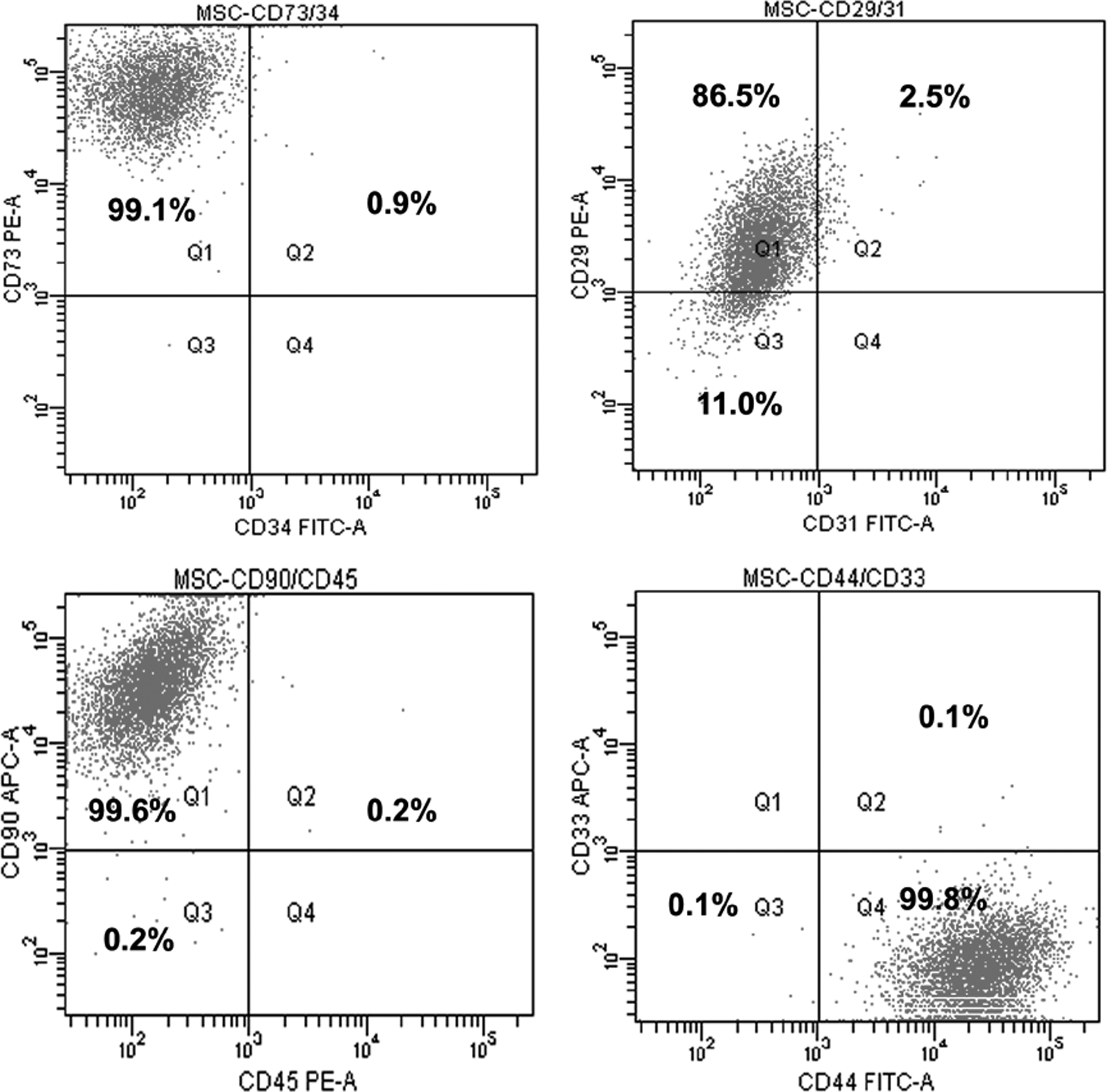
Dual flowcytometric analysis of UCB-hMSCs after passage 3 for MSC specific surface markers such as CD44, CD73, CD90, and CD29 against the lineage specific markers CD31 (endothelial cells), CD33 (myeloid cells), CD34 (hematopoetic cells), and CD45 (leucocytes). The flow cytometric histograms of unstained negative control for each fluorochrome combinations are given as Supplementary data (Supplementary Fig. S1). UCB-hMSCs, human umbilical cord blood-derived mesenchymal stem cells.
Cell attachment and spreading
The SEM micrographs (Fig. 8) showed that the UCB-hMSCs adhered well, started to spread on the electrospun fibrin (ES-Fibrin) scaffold within 6 h of cell seeding, and covered the scaffold uniformly by 48 h. The cells were found to be very sensitive to the scaffold composition, which can be accredited to the integrin binding sites found on fibrin such as Arg-Gly-Asp and Ala-Gly-Asp-Val, through which it can interact with the cell surface integrins. 11

SEM image of UCB-hMSCs grown on ES-fibrin scaffold for 6 h
In correlation with the above data, the UCB-hMSCs adhered on the ES-fibrin scaffold over a 24 h period showed good F-actin organization (Fig. 9). This organization is thought to be dictated by the biofunctionality imparted by the nano fibrous fibrin scaffold. In vitro, cells adhere to the underlying matrix through different mechanisms, which include ECM contacts, close contacts, and focal adhesions. Adhesive interactions play an important role in regulating many cellular functions during development and remodeling. Cell architecture is controlled by many additional factors, including actin, myosin, microtubules, and extrinsic extracellular and mechanical forces. The actin cytoskeleton directs adhesion assembly, disassembly, and organization. The physical linkage through which the integrins and actins are bonded together provide a grip for migration and crucial for the integrity and stability of adhesions. Integrins are a family of cell surface receptors at discrete sites called “focal adhesions” through which cell adhere to the ECM. 38 Integration of the signals that causes the rearrangement of the actin cytoskeleton takes place in the focal adhesions. 39
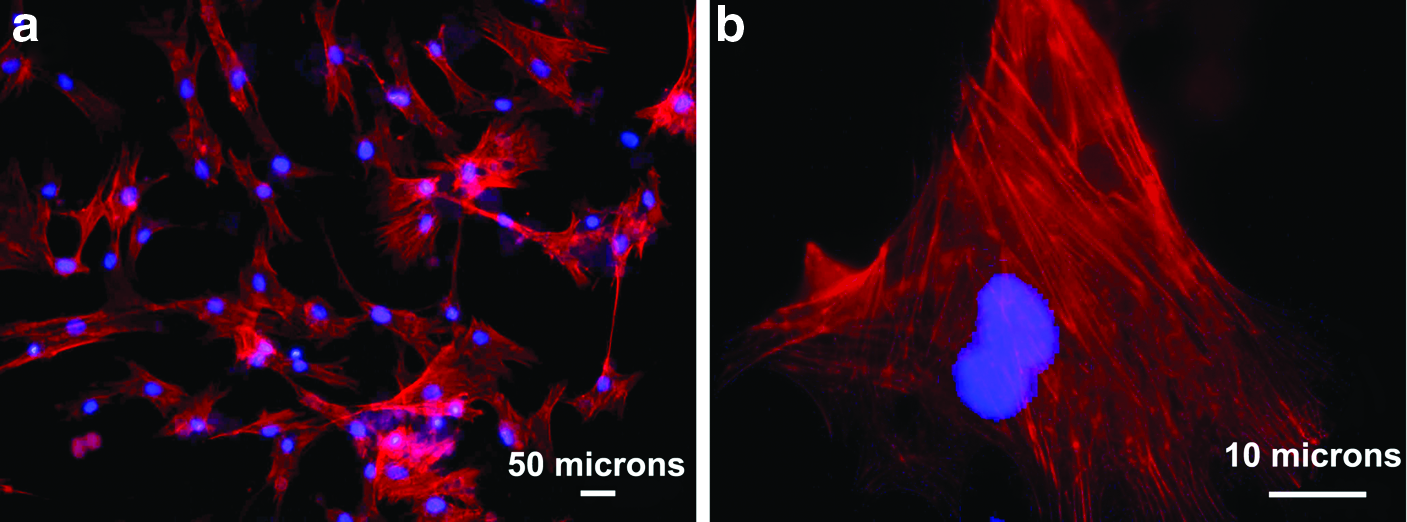
Merged fluorescent images of F-Actin filaments (Red) stained with TRITC-conjugated phalloidin and DAPI-stained nuclei (blue) of cells grown ES-Fibrin for 24 h at
In our study, in addition to the well-organized actin filaments, the cells were found to spread and arrange their cytoskeletons with large “dash” like focal adhesions on the scaffold within 24 h. Interestingly, Vinculin were localized in dash-like focal adhesion complexes that were clearly associated with actin filaments. Focal adhesions appeared yellow, when the fluorescent images were merged, indicating inclusive colocalization of vinculin with actin fibers (Fig. 10c, d). Fath et al. reported that the accumulation of specific integrins in focal contacts depends on the composition of the substratum. 40 Overall, the well-defined actin stress fibers and focal adhesion complexes provided tight binding to the underlying fibrin rich ECM. A relatively weak interaction between integrins and their substrate ligands would have resulted in an inefficient coupling, as would the interaction with ligand on a pliable substrate.

Confocal microscopy images of UCB-hMSCs grown on ES-Fibrin over a period of 24 h. Fibrin promote the formation of stress fibers and focal adhesions that contain vinculin. Upper panel shows well defined “dash like focal adhesions” as indicated by the presence of vinculin (green). Nuclei are stained with PI (Red). Focal adhesions that appear yellow in the lower panel indicate the regions of actin-vinculin colocalization. Enlarged portions of the image are shown in the inset. Color images available online at
Cell proliferation
Good retention and viability of the cells are critical issues for the clinical use of tissue-engineered constructs. The evaluation of the retention of the hMSCs on the scaffold in vitro can present a preliminary authentication of the applicability of this novel ES-fibrin scaffold. The proliferation of cells on the scaffold was evaluated by Alamar blue assay and nuclear staining using DAPI. Figure 11 depicts the proliferation of MSCs on the scaffold in comparison with that on the gold standard, tissue culture polystyrene and fibrin-coated cover slips. All data were normalized to the initial seeding density at day 0, as assessed 6 h after seeding. It is evident from Figure 11 that the cell proliferation is significantly better on ES-fibrin scaffold, which may be attributed to the topography and biological nature of nanofibrous fibrin.

Fluroescence image of the DAPI-stained nuclei of UCB-hMSCs grown on polystyrene cover slips
Conclusion
This study reports a novel electrospinning method for the fabrication of nano fibrous fibrin scaffold through a biomimetic route, capable of supporting in vitro adhesion and proliferation of UCB-hMSCs. Our results demonstrate that the average diameter of the ES-fibrin fibers could be scaled down to 50–500 nm by electrospinning, which exactly mimics the natural ECM in dimension. Also, the use of plasma-derived fibrinogen rich cryoprecipitate containing factor XIII eliminates the need of a chemical cross-linking agent that can be deleterious to the bioactivity of the scaffold. The most striking advantage of this novel electrospinning method over other conventional coating techniques is that a uniform distribution of fibrin with predictable fiber morphology and dimension can be obtained, which promotes the efficient cellular infiltration throughout the scaffold. Moreover, the cell-interaction studies with UCB-hMSCs reveals the application potential of our scaffold in various avenues of tissue engineering. For soft tissues such as skin, nerve, cornea, and so on, this scaffold can be used as such, but for hard tissues that need high mechanical strength, the structural and mechanical integrity of the fibrin nanofibers might not be sufficient. In such cases, synthetic polymers such as PCL, PLCL, PLGA, and so on can be used in combination with fibrin to produce composite tissue-engineered scaffolds with tunable mechanical property according to tissue-specific needs/applications.
Footnotes
Acknowledgments
The authors are grateful to the Department of Science and Technology, the Government of India, for funding this work through the National Nanoscience and Nanotechnology Initiative monitored by Professor C. N. R. Rao. The authors gratefully acknowledge the help from Mr. Sajin P Ravi for SEM analysis.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.