
Abstract
Mesenchymal stromal cells (MSC) represent a promising population for supporting new clinical concepts in cellular therapy. A wide diversity of isolation procedures for MSC from umbilical cord blood (UCB) has been described for humans. In contrast, a few data are available in horses. In the current study, a sedimentation method using hydroxyethyl starch and a method based on the lysis of red blood cells using ammonium chloride (NH4Cl) were compared with two density gradient separation methods (Ficoll-Paque and Percoll). Adherent cell colonies could be established using all four isolation methods. The mononuclear cell recovery after Percoll separation, however, resulted in significantly more putative MSC colonies; and, therefore, this isolation method was used for all further experiments. Culture conditions such as cell density and medium or serum coating of the wells did not significantly affect putative MSC recovery. Isolated MSC using Percoll were subsequently differentiated toward the osteogenic, chondrogenic, and adipogenic lineage. In addition, MSC were phenotyped by multicolor flow cytometry based on their expression of different cell protein markers. Cultured MSC were CD29, CD44, and CD90-positive and CD79α, Macrophage/Monocyte and MHC II-negative. In conclusion, this study reports optimized protocols to isolate, culture, and characterize solid equine MSC from UCB.
Introduction
Several UCB fractionation procedures for the isolation of human MSC have been proposed based on the partial or complete removal of red blood cells (RBCs) and plasma. 12 These include (i) the use of sedimentation agents such as hydroxyethyl starch (HES),13,14 poligeline 15 or gelatin,16,17 (ii) simple manual or partially automated centrifugation, 18 and (iii) density gradient cell separation protocols based on either Percoll19,20 or Ficoll.3,21,22–26 In human medicine, few reports compare the efficacy of different isolation methods in terms of cell recovery.12,15,17,27 In veterinary medicine, two studies have been published to date comparing different isolation protocols for equine MSC that had been derived from equine bone marrow 28 or from UCB, 29 respectively.
The major aim of this study was to compare four isolation methods to acquire putative equine MSC from UCB: one method based on the lysis of RBC using NH4Cl, one sedimentation method with HES, and two methods based on the use of density gradient separation that is, Percoll and Ficoll-Paque. In addition, the importance of culture conditions such as cell density, culture medium, and serum coating of the wells for in vitro culturing of putative UCB-derived equine MSC was evaluated. A final objective was to characterize the putative isolated MSC both by assessment of their differentiation capacities and by immunophenotyping.
Materials and Methods
UCB collection
UCB was collected from full-term born foals immediately after birth, before the umbilical cord spontaneously ruptured. After clamping and disinfecting the umbilical cord with 70% alcohol, the umbilical vein was punctured, and UCB was drained by gravity into a sterile standard 350-mL blood donor bag containing 49 mL CPD A anticoagulant (Terumo®), and subsequently stored at 4°C. A number of critical conditions, as described by Bieback et al., 3 were included to decide whether further processing of the UCB was attempted. As such, the volume of all samples processed was more than 100 mL (197.1±39.0 mL), storage time was less than 15 h (9.8±3.0 h), and none of the samples showed signs of coagulation or hemolysis.
Cell isolation methods
Eight UCB samples were collected in order to compare the four different isolation methods
To fit the experimental design (see below), 2.8×107 isolated cells were required. To this end, different volumes of UCB were processed for each isolation method, that is, 4 mL for NH4Cl, 60 mL for Percoll, 120 mL for Ficoll-Paque, and 40 mL for HES.
Lysis of the RBC with NH4Cl lysing solution
Fourteen mL NH4Cl lysing solution (consisting of 8.9 g NH4CL, 1.0 g KHCO3, 37.0 mg tetrasodium ethylenediaminetetraacetic acid (EDTA) in 1 L distilled water, all from Sigma, pH=7.3) was added to a 15 mL Falcon tube containing 1 mL UCB at room temperature (RT). During 3 to 5 min, four tubes were inverted several times and subsequently centrifuged for 5 min at 300 g at RT. After removing the supernatant, the cell pellet was washed in Hank's balanced salt solution without Ca/Mg (HBSS) (Invitrogen) by centrifuging 5 min at 300 g, and finally resuspended in 5 mL HBSS.
Density gradient separation with Percoll
After centrifuging 60 mL UCB at 1000 g for 20 min at RT, the buffy coat fraction was collected and diluted 1:1 (v:v) with HBSS. Subsequently, the cell suspension was gently layered on an equal volume of Percoll (density 1.080 g/mL; GE Healthcare) and centrifuged for 15 min at 600 g at RT, as previously described. 19 The interphase was collected, washed thrice with HBSS by centrifuging 10 min at 200 g, and finally resuspended in 5 mL HBSS.
Density gradient separation with Ficoll-Paque
The mononuclear cell fraction was isolated by loading 30 mL of UCB onto 10 mL Ficoll-Paque PREMIUM® (GE Healthcare) in 50 mL Falcon tubes, as described by Koch et al. 30 Four tubes were centrifuged for 30 min at 450 g at RT. After aspirating the supernatant, the interphase was collected and washed twice with HBSS by centrifuging 5 min at 450 g, and finally resuspended in 5 mL HBSS.
HES separation
HES solution (HES 6%®; Braun) was added to UCB in a ratio of 1:5 (v:v). After centrifugation of four tubes for 5 min at 50 g at RT, the leukocyte-rich plasma, containing some RBC, was collected. This fraction was centrifuged again for 15 min at 600 g at RT. The supernatant was subsequently removed, and the cell pellet was washed with HBSS by centrifuging 10 min at 200 g, and finally resuspended in 5 mL HBSS.
Media
Two different culture media were compared for the isolation and expansion of MSC from UCB. MesenCult® is a commercial MSC medium based on McCoy's medium (StemCell Technologies). The other culture medium used was largely based on the medium described by Koch et al., 30 hereafter designated Koch's medium, and contains low-glucose Dulbecco's Modified Eagle Medium (DMEM; Invitrogen), 30% fetal calf serum (FCS; GIBCO), 10−7 M low dexamethazone, 50 μg/mL gentamycine, 10 μL/ml antibiotic antimycotic solution, 250 ng/mL fungizone (all from Sigma), and 2 mM ultraglutamine (Invitrogen). Expansion medium was identical to Koch's culture medium but without dexamethasone.
For the differentiation experiments, the following media were used: (i) osteogenic medium, containing low glucose DMEM (Invitrogen), 10% FCS (GIBCO), 0.2 mM L-ascorbic acid-2-phosphate (Fluka), 100 nM dexamethasone, 10 mM β-glycerophosphate, 50 μg/mL gentamycine, and 10 μL/mL antibiotic antimycotic solution (all from Sigma); (ii) chondrogenic medium based on the basal differentiation medium (Lonza), complemented with 10 ng/mL Transforming Growth Factor β3 (Sigma) and (iii) adipogenic induction medium containing DMEM-LG (Invitrogen), 1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 10 μg/mL rh-insuline, 0.2 mM indomethacin, 15% rabbit serum, 50 μg/mL gentamycine, and 10 μL/mL antibiotic antimycotic solution (all from Sigma); (iv) adipogenic maintenance medium, which was identical to the adipogenic induction medium except for the omission of dexamethasone, indomethacin, and 3-isobutyl-1-methylxanthine.
Experimental design
After isolation, cells were seeded at three different cell concentrations (1×106, 2×106 and 4×106 cells/mL) in two different media (Mesencult and Koch's medium) using 2 mL medium per well. Moreover, isolated cells were seeded on uncoated polystyrene six-well culture dishes (BD) as well as on polystyrene six-well culture dishes coated with 100% FCS (GIBCO). As a result, UCB-derived blood cells from one mare were isolated using four different methods, and 12 different culture conditions were evaluated for each isolation method. So, each UCB sample was subdivided in 48 units with one unit representing one tested condition (Fig. 1).
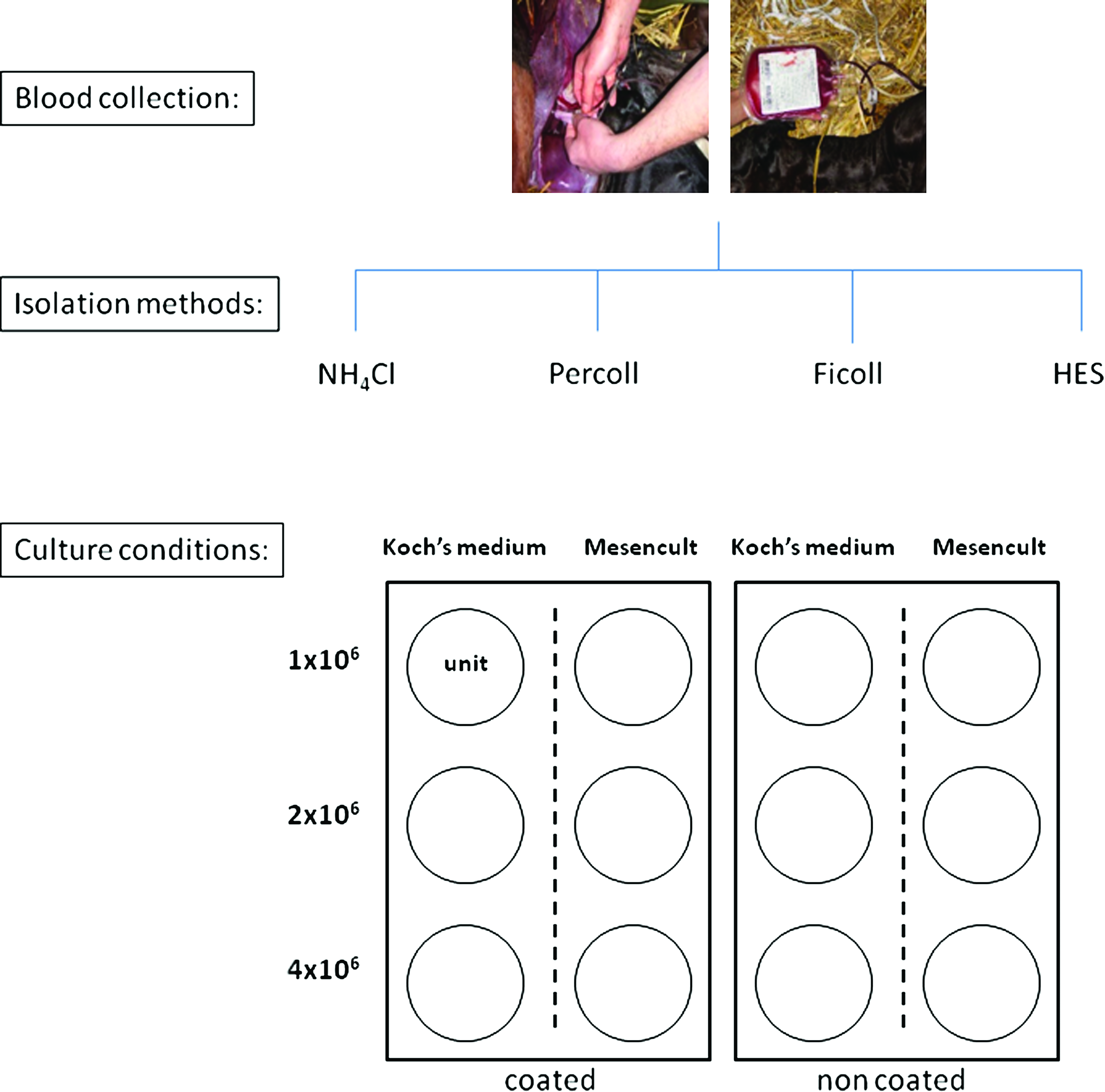
Schematic overview of the experimental design. For each umbilical cord blood sample, blood cells were isolated using four different methods and for each isolation, 12 different culture conditions were tested such as culture medium, seeding concentration, and coated versus noncoated wells. Each condition is represented by one unit. HES, hydroxyethyl starch. Color images available online at
Culture parameters and conditions
Cell viability was determined by trypan blue exclusion using the improved Neubauer hemocytometer, as previously described. 31 Isolated cells were incubated at 38.5°C in a humidified atmosphere containing 5% CO2. After overnight incubation, nonadherent cells were removed, and fresh medium was added to the wells. The remaining nonadherent cells were removed by exchanging the culture medium every seven days. Cultures were inspected every three days for the formation of adherent spindle-shaped fibroblastoid cell colonies. Cells were passaged as soon as confluency exceeded 80% using 0.083% trypsin-EDTA (Sigma). The replating ratio after chemical cell detachment was 1:3.
Tri-lineage cell differentiation
Mononuclear cells from seventeen UCB samples were isolated using Percoll and cultured at a concentration of 4×106 cells/mL in uncoated T-25 culture flasks using Koch's medium. The medium was exchanged every 3–4 days. Cells were subsequently expanded in expansion medium for the next two passages. All experiments were performed in triplicate, and noninduced cells in expansion medium were used as negative controls.
Osteogenic differentiation
The osteogenic differentiation was performed in six-well culture dishes with approximately 3000 cells/cm2 that had been were cultured in expansion medium until 90%–100% confluency was reached. Hereafter, osteogenic differentiation was induced with osteogenic medium that was changed every 3–4 days. Osteogenic differentiation was evaluated after 20 days of culture using the Alizarine Red S and the Von Kossa histological staining after fixation with 4% buffered formaldehyde, as well as by detecting alkaline phosphatase activity (Millipore®, Alkaline Phosphatase Detection kit), according to the manufacturer's instructions.
Chondrogenic differentiation
Approximately 2.5×105 cells were centrifuged in 15-mL conical Falcon tubes (150 g for 5 min at RT), whereafter 0.5 mL chondrogenic medium was added to each tube without disturbing the cell pellet. These micromass culture systems were maintained for 3 weeks, replacing the medium every 3–4 days. After fixation with 4% buffered formaldehyde overnight, the pellets were embedded in 2% agarose and further processed for routine paraffin sectioning. Chondrogenic differentiation was evaluated by the Alcian blue histological staining on 8 μm thick sections.
Adipogenic differentiation
Approximately 2.1×104 cells/cm2 were seeded in six-well culture dishes and cultured until 100% confluency was reached. Cells were then exposed to four cycles of 72 h culturing in the adipogenic induction medium and 24 h of culturing in the adipogenic maintenance medium, followed by five consecutive days of culturing in adipogenic maintenance medium. 30 After 21 days, adipogenic differentiation was assessed using Oil Red O histological staining after fixation in 4% buffered formaldehyde.
Flow cytometry
After Percoll isolation, mononuclear cells were cultured and subsequently, the obtained MSC were expanded for the next three passages and used at a concentration of approximately 2×105 cells per tube. In general, cells were washed in DMEM+1% BSA and for intracellular antigen detection (CD79α and macrophage/monocyte marker), first fixed and permeabilized using Fix en Perm® (Caltag; Invitrogen) according to the manufacturer's instructions. Cells were then incubated for 15 min at 4°C in the dark with combinations of either unlabeled or directly fluorescent-labeled monoclonal antibodies (mAbs) to obtain a multicolor analysis of the markers. The mAbs used in this study were directed against human CD29 (BioLegend, 309016), human CD44 (BioLegend, 559250), canine CD90 (VMRD, Inc., DH24A), equine MHC-II (MCA1085), human CD79α (MCA2538A6), and a human macrophage/monocyte marker (MCA874A48) (all from Serotec). As positive controls, freshly isolated equine peripheral mononuclear cells and endothelial cells were used to test for species cross-reactivity; and as negative controls, cells were incubated either with secondary Ab alone or with isotype controls: rat IgG2 (BioLegend, 400611) for CD29, mouse IgG1 (BioLegend, 400132) for CD44, and mouse IgM (Becton Dickinson, 557275) for CD90, respectively. After two washing steps, cells that had been incubated with nonlabeled mAbs were subsequently incubated with a secondary Ab for 15 min at 4°C in the dark. Secondary Abs used were R-Phycoerythrin-conjugated sheep anti-mouse IgG (Sigma, P8547) and Alexa 647-conjugated goat anti-mouse IgG (Invitrogen, A21235). Cell pellets were finally washed twice to remove the excess of secondary Ab and resuspended in 400 μL PBS. At least 10,000 cells were analyzed using the FACScanto flow cytometer (Becton Dickinson Immunocytometry systems) equipped with two lasers, a 488 nm solid state and a 633 nm HeNe lazer, and FACSDiva software. All data were corrected for autofluorescence using autofluorescent tubes, as well as for unspecific bindings using secondary Ab negative controls and isotype controls.
Statistical analysis
Data were presented as median±interquartile range. Kruskal–Wallis tests were performed to evaluate whether the isolation method used was associated with concentration and viability. Cox proportional hazard survival models were fit to study the association between growth of cells (1=growth, 0=censored) and different predictor variables (cell isolation method, cell concentration, culture medium, and serum coating). Mare was forced into the model to correct for clustering. A multivariable model was built omitting nonsignificant (p<0.05) variables using a backward stepwise approach. Hazard ratios (HR) with 95% confidence intervals were calculated. A Kaplan-Meier graph was generated. All analyses were performed using SPSS 16.0 (SPSS Inc. Headquarters).
Results
Isolation of putative MSC using Percoll gradient centrifugation results in the highest number of adherent spindle-shaped cell colonies
Eight UCB samples were collected without any complication, and blood cells from each sample were isolated in parallel. The cell viability was comparable for the four isolation methods (p=0.15) (Table 1). Generally, adherent spindle-shaped cell colonies occurred within 10.1±4.4 days (ranging from 6 to 32 days), and 80% to 100% cell confluency was reached after 15.6±1.8 days of culture (Fig. 2). Interestingly, colony formation of UCB-derived blood cells was significantly influenced by the mare (e.g., age, parity, genetic background,…) on the one hand (p<0.001) and the isolation method used on the other hand (p<0.001) (Table 2). For the latter, it was found that adherent spindle-shaped cell colonies were formed in 37 out of 96 units (38.5%) by means of the Percoll method, whereas this was only in 15 out of 96 units (15.6%) when using Ficoll-Paque (HR=0.262) (Table 2). For both the NH4Cl and HES method, adherent colonies were observed in only 1 out of 96 (1.0%) units (HR=0.013 and 0.017 respectively) (Table 2). The first colonies were recovered as early as six days after the start of culturing Percoll-isolated mononuclear cells and from ten days onwards, larger adherent spindle-shaped cell colonies were formed as shown by the clear increase in the percentage of units showing growth (Fig. 3). A similar pattern was also observed when culturing Ficoll-isolated mononuclear cells, but to a much lower extent (Fig. 3). Almost no growing spindle-shaped cell colonies were observed with blood cells isolated by means of either NH4Cl or HES (Fig. 3).
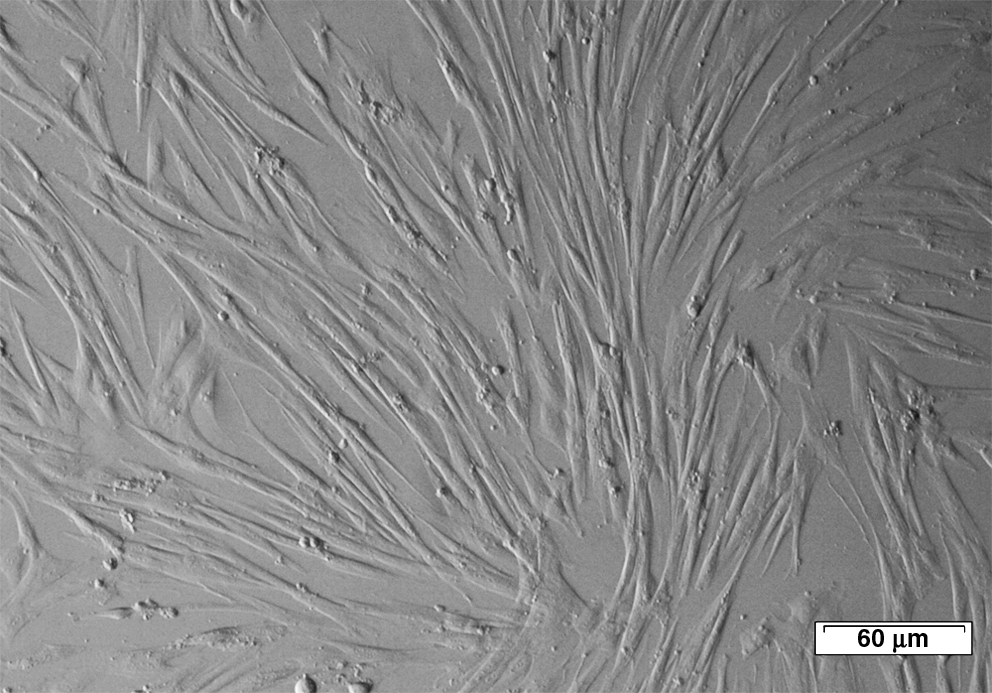
Representative light microscopy picture of spindle-shaped adherent MSCs. Scale bar: 60 μm. MSC, mesenchymal stromal cell.
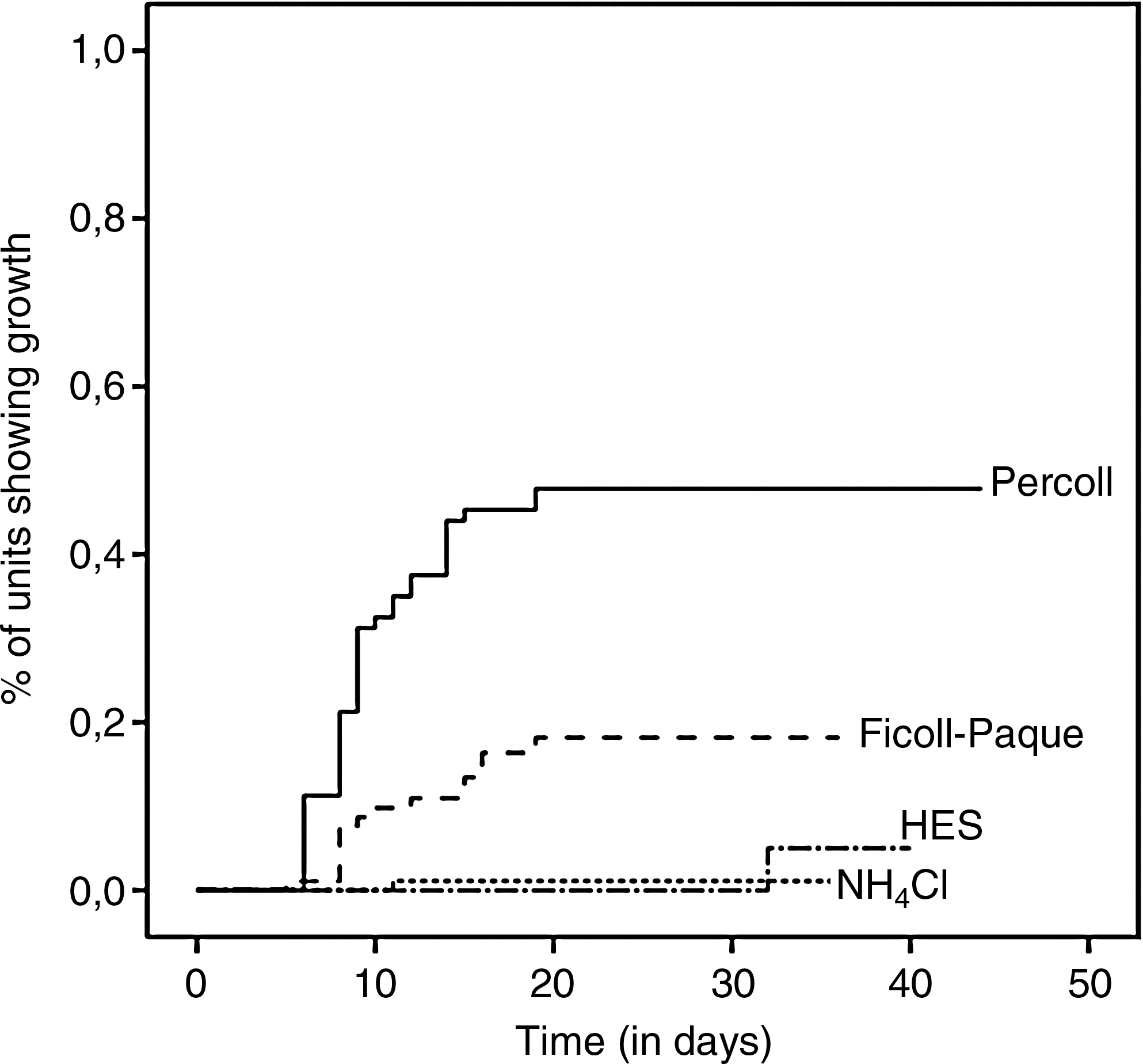
Growth of putative MSC units. Kaplan-Meier graph showing growth over time (days) in function of the four isolation methods used.
HES, hydroxyethyl starch; UCB, umbilical cord blood.
95% CI around HR.
Data for individual mares are not shown in this table.
Reference category.
HR, Hazard ratio; 95% CI, 95% confidence interval; NS, nonsignificant.
In contrast to the significant influence of the isolation method used, colony formation was not influenced by the original seeding concentration, nor by the absence or presence of coating, nor by the culture medium used (Table 2). When blood cells were cultured at a concentration of 4×106 cells/mL, colonies were noted in 19 out of 128 units (14.8%), which was not significantly different from the results obtained with the concentrations of 2×106 cells/mL and 1×106 cells/mL (16 and 19 out of 128 units (12.5% and 14.8%), respectively) (Table 2). Culturing isolated blood cells in commercial MesenCult medium or Koch's medium resulted in similar numbers of units showing growth, namely 27 out of 192 units (14.1%) for both culture media (Table 2). Finally, seeding blood cells in FCS-coated wells resulted in growing colonies in 29 out of 192 units (15.1%), which was not significantly different from the growth rates obtained when cells were seeded in uncoated wells (25 out of 192 units [13%]) (Table 2).
UCB-derived putative MSC are capable of differentiation
Putative MSC were isolated in 13 out of 17 UCB samples with the optimized isolation and culture protocol as just described. In these samples, adherent spindle-shaped cell colonies started to grow within 9.9±2.2 days (ranging from 7 to 14 days) and 80% cell confluency was reached after 15.5±2.2 days of culture.
From five UCB samples, putative MSC at their third passage were used to initiate in vitro osteogenic, chondrogenic, and adipogenic differentiation. It was found that the cells were able to differentiate into osteocytes, as demonstrated by an increased alkaline phosphatase activity compared with the negative control group (Fig. 4A, D). In addition, phosphate and calcium deposits, which are osteogenic specific features, were demonstrated using Alizarine Red S and Von Kossa staining, respectively (Fig. 4B, C, E, F). Differentiation toward the chondrogenic lineage was confirmed by a positive Alcian blue staining (Fig. 5A–C), which identifies the acid mucins in the chondrogenic matrix. Differentiation of the MSC toward adipocytes was confirmed by a positive Oil Red O staining, which is used to detect the intracellular accumulation of lipid droplets (Fig. 6A, B).
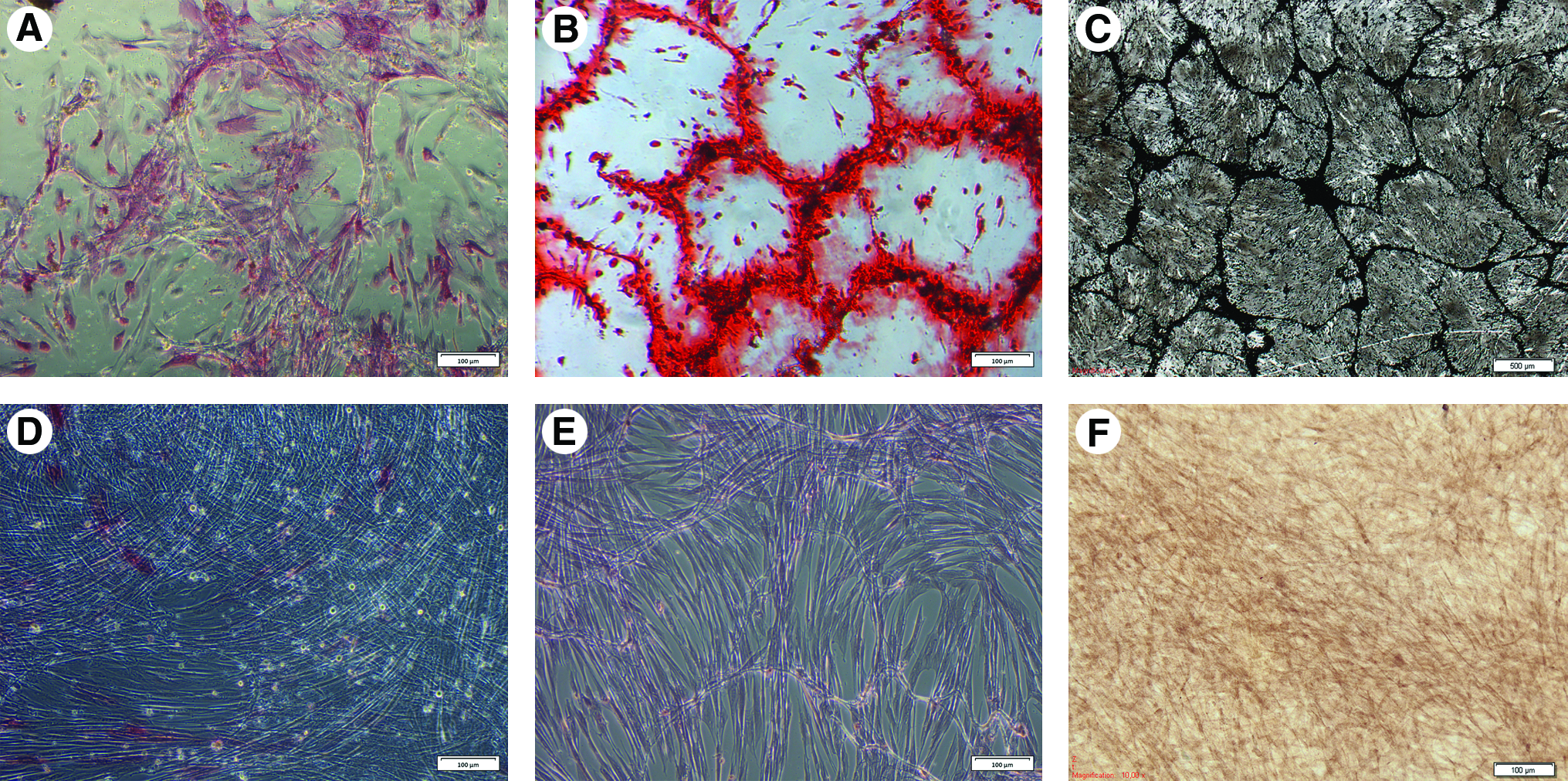
MSCs can differentiate into osteocytes. Differentiation toward the osteogenic lineage was confirmed by increased alkaline phosphatase activity
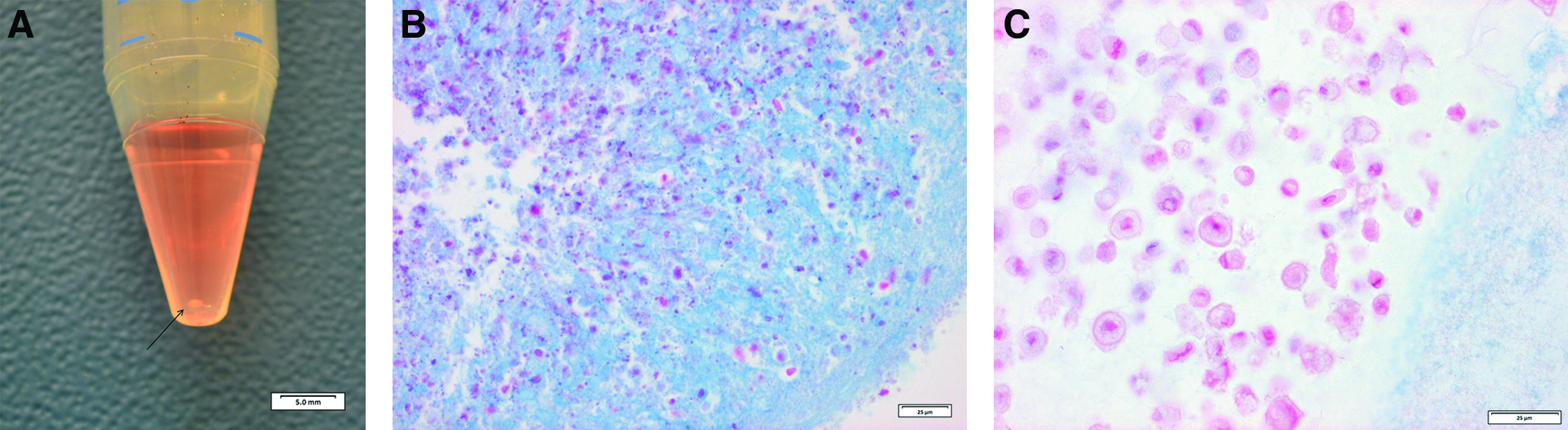
MSCs can differentiate into chondrocytes. Differentiation toward the chondrogenic lineage was performed using a micromass culture system

MSCs can differentiate into adipocytes. Differentiation toward the adipogenic lineage was confirmed by Oil Red O
UCB-derived putative MSC exhibit required immunophenotypical characteristics
An important feature to identify solid MSC is their immunophenotyping based on the expression of a set of cell markers such as CD29, CD44, CD73, CD90, and CD105, and the absence of other cell markers that are mainly present on leukocytes and hematopoietic stem cells, that is, CD34, CD45, CD11b or CD14, CD19 or CD79α, and MHC-II.32,33 To this end, putative MSC from the 4th passage were used and incubated with a mixture of mAbs against human CD29, human CD44, canine CD90, equine MHC-II, human CD79α, and a human macrophage/monocyte marker. Hereby, it was found that the equine MSC expressed CD29, CD44, and CD90 (>90%) and lacked expression of CD79α, the macrophage/monocyte marker, and MHC II (<5%) (Fig. 7). Expression of CD34, CD45, CD73, and CD105 could not be evaluated, as no cross-reactivity could be demonstrated for these mAbs using the proper equine control cells (data not shown). Isolated MSC were cryopreserved, thawed, and cultured for one passage, whereafter they were flow cytometrically characterized again. No significant differences were observed between these cryopreserved and fresh MSC (data not shown).

Immunophenotype of equine MSCs. Isolated MSC were positive for CD29
Discussion
Isolation of human MSC from UCB has been described with varying success.3,21,22,34–37 The isolation step could be the main cause for this variation in outcome, as this factor has been reported to be critical when obtaining equine MSC from bone marrow. 28 Therefore, the major aim of the present study was to evaluate different isolation procedures to obtain putative MSC from equine UCB. Each UCB sample was subjected to four different isolation methods, which is a more favorable experimental set-up than the random allocation of samples to one of the different procedures, because the influence of variation between UCB collections is avoided. Two methods of density mononuclear cell fractionation, namely Percoll and Ficoll-Paque, were included, as these techniques are regularly used to obtain human MSC.3,19–25 In addition, we included two techniques to obtain the whole white blood cell fraction instead of only the mononuclear cell fraction to start the MSC culture. RBCs were chemically lysed using NH4Cl, which has been described as a reliable and effective alternative to density-gradient centrifugation. 38 On the other hand, rouleaux formation of RBC induced by HES, first described by Rubinstein et al. 13 for human UCB, was also included.
The proliferation of MSC varied significantly between mares, which can be partly explained by differences in storage time and UCB volume, as has been suggested in other studies.14,18 Putative equine MSC could be obtained when the mononuclear cell fraction was isolated using both density gradient separation methods. On the other hand, adherent cell colonies were only observed on a single occasion when HES and NH4Cl were used, which is in contrast to the report that used NH4Cl for the isolation of human MSC from bone marrow. 38 Possible explanations for our lack of success to culture putative MSC after HES or NH4Cl isolation could be the presence of polymorphonuclear cells such as neutrophils and the contamination with remaining RBC, as these factors have been reported to decrease cell adherence and proliferation.27,39
When comparing both density gradient isolation methods, a significantly higher number of adherent colonies was observed with Percoll in comparison to Ficoll-Paque. In addition, the percentage of colonies that subsequently proliferated was much higher. Interestingly, Percoll is not commonly used to isolate MSC from equine UCB.28,40–42 The reason that the frequently used Ficoll-Paque based isolation technique was not as successful in recovering putative equine MSC in the current study varies. First, in the Percoll isolation protocol used, the UCB was first centrifuged to obtain the buffy coat fraction in contrast to the Ficoll Paque isolation, where whole blood was used. In humans, the buffy coat fraction has been described to contain almost 70%–90% of human stem cells. 43 Further, the difference in chemical composition of the two gradient media may contribute to the variation in outcome of the isolation of equine putative MSC from UCB. Ficoll-Paque is based on a mixture of a synthetic sucrose polymer and an iodinated compound, whereas Percoll consists of colloidal particles coated with polyvinylpyrrolidone. 44 The main disadvantages of sucrose solutions, such as Ficoll, are their physico-chemical properties including a high osmolality and viscosity, 45 although the iodinated compound in the Ficoll-Paque used in the present study should have eliminated the high osmotic stress. 44 Still, Percoll is less viscous implying a reduced risk of cell agglutination. 46 Further, Percoll does not alter the density of monocytes, which results in a better separation of lymphocytes and monocytes.46,47
Apart from the isolation methods, the influence of some culture parameters such as seeding density, culture media, and coating of the wells with FCS was evaluated. Reported seeding densities of blood cells for MSC isolation broadly range from 1×104 cells/cm2 up to 1×106 cells/cm2.3,20,48,49 A higher seeding density will lead to a higher secretion of biologically active factors by the plated cells, which may contribute to cell survival as well as angiogenesis.50,51 On the other hand, the seeding density cannot be too high either, because nonadherent cells as well as noncellular debris can possibly block an effective attachment of the target cells to the plastic. No significant differences were found in the recovery of putative equine MSC in the current study, thus indicating that a seeding concentration from 1 up to 4×106 cells/ml is a good range for a proper equine MSC isolation from UCB. In addition, we did not find significant differences in putative equine MSC recovery between (i) the seeding of cells on uncoated versus FCS-coated wells and (ii) the two culture media tested. Coating with FCS was evaluated in our study, because it has been used in human MSC cultures to prevent stable adherence of monocytic cells. 3 Indeed, Bieback et al. 3 observed a significantly higher percentage of monocytes in the nonadherent fraction after plating the mononuclear cell fraction on FCS-coated wells, which indicates that fewer monocytes adhere to coated plates in comparison to uncoated plates. In contrast to the latter paper but in accordance with our findings, Koerner et al. 1 reports variable results when wells were pre-coated with FCS. Since there is the factor of variation between different batches of FCS, we decided to use noncoated wells for culturing equine MSC in the optimized protocol. Finally, two different media that is, MesenCult and Koch's medium, were tested, because previous research indicated that human bone-marrow derived MSC could be cultured using MesenCult, whereas attempts to isolate human MSC from UCB using this medium failed. 3 In the current study, culturing putative MSC was successful using either medium, indicating that the medium is not a determining factor for the isolation of putative equine MSC from UCB.
Apart from the differentiation experiments, the UCB-derived equine MSC were also immunophenotypically characterized. We found that the isolated cells expressed CD29, CD44, and CD90, and were negative for CD79α, MHC II, and a macrophage/monocyte marker. In general, the phenotypical identification of equine MSC is hampered by the limited availability of species-specific or cross-reacting mAbs. In the current study, a macrophage/monocyte marker was used that detects calprotectin, an intracellular protein with a restricted distribution within the monocyte-derived cell lineage. 52 This mAb was chosen instead of mAbs recognizing CD11b or CD14, because the cross-reactivity of this mAb with equine calprotectin had already been confirmed, 53 in contrast to the cross-reactivity potential of mAbs recognizing human CD11b or CD14 at the time of these experiments.
In conclusion, we describe in the current study an optimized protocol for isolating and culturing equine MSC from UCB. Importantly, the isolated equine MSC were in addition adequately characterized by assessing their differentiation capacities and immunophenotypic features. Studies comparing different isolation methods in veterinary relevant species such as the horse are of added value to the field, as Ficoll-Paque is the most commonly used density medium to isolate MSC, whereas the current study clearly shows that Percoll gives a significantly better yield. Equine UCB can be considered a good and reliable source for solid equine MSC, which could have a major importance in the growing field of veterinary cell-based therapies.
Footnotes
Acknowledgments
The authors gratefully acknowledge Petra Van Damme, Isabel Lemahieu, and Leen Pieters for their excellent technical assistance. Jurgen De Craene has performed the histological stainings. Thomas Koch is thanked for his useful advice.
Disclosure Statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.