
Abstract
Replacement of a diseased organ with an autologously derived tissue is an ideal therapy for some medical problems. However, it is difficult to recreate many adult human tissues in vitro due to the functionally necessary architecture of most organs and the lack of understanding of methods to direct the development of the organ of interest. The parathyroid gland is ideal for in vitro organ development because this gland is relatively simple, is transplantable, and is commonly affected by a surgical complication rather than an autoimmune disease. We have investigated thymus as a source of autologous endoderm and parathyroid-like precursor cells. Human thymus cells were treated with a differentiation protocol we developed with human embryonic stem cells (The Bingham Protocol) that utilizes timed exposures to Activin A and soluble Sonic hedgehog (Shh). We incrementally changed the protocol to optimize the differentiation of the thymus cells into parathyroid-like cells. The final protocol used 50 ng/mL Activin A and 100 ng/mL Shh over 13 weeks. The differentiated cells expressed the parathyroid markers parathyroid hormone (PTH), calcium sensing receptor, chemokine receptor type-4 (CXCR4), and chorian-specific transcription factor (GCM2) as measured by reverse transcription–polymerase chain reaction and PTH enzyme-linked immunosorbent assay. Cultured thymus cells without Activin A or Shh exposure did not secrete PTH nor express similar markers. The differentiated cells released PTH, which was suppressed in response to increased calcium concentration. The chemically differentiated cells did not form tumors in immune-compromised mice. Our protocol recreated cells with markers of parathyroid tissue that responded as parathyroid cells to physiologic stimuli. This approach is a further step toward a strategy to restore parathyroid function using autologous cells that were directed to differentiate by nongenetic in vitro manipulation.
Introduction
The four parathyroid glands are small, delicate structures that share their blood supply with the thyroid gland. Parathyroid cells detect and regulate serum calcium levels. Because the parathyroid glands can be difficult to identify in situ, accidental loss of parathyroid gland function (hypoparathyroidism) is the most frequent permanent complication of thyroid and parathyroid surgery. In current clinical practice, devascularized parathyroid tissue can be mechanically disrupted into small bits and inserted into tissue pockets where the cells survive on diffused nutrients until neovascularization occurs. Resumption of measurable, normal parathyroid function occurs in 6–10 weeks postgrafting for patients who otherwise have no endogenous parathyroid function. The rate of complete normalization of parathyroid function after autograft of parathyroid tissue is 50%–95%, 1 and the grafted parathyroid tissue responds normally to changes in serum calcium concentration. Unfortunately, unless damage to the parathyroid glands is recognized in the operating room, autografting cannot be performed.
Even though prevention of hypoparathyroidism by autografting damaged parathyroids is widely recognized as the best approach, the rate of permanent hypoparathyroidism after thyroid surgery is nearly 14% in population-based series2–4 though the occurrence is much lower in series from expert surgeons (0.5%–4.0%).5–7 The chronic, detrimental effects of hypoparathyroidism on bone, teeth, skin, and nails are well documented. 8 Hypoparathyroidism results in chronic hypocalcemia and a low-turnover bone disease that are poorly managed by currently available replacement methods, which include multiple daily doses of vitamin D analogues and calcium salts. Replacement of parathyroid hormone (PTH) itself is available as teriparatide (Forteo, Lilly), a 1–34 N-terminal protein fragment of PTH; however, the serum half-life of synthetic PTH is less than 5 min. While the overall treatment plan for hypoparathyroidism can be effective, studies have shown that treatment regimens with multiple daily doses, with drugs that can cause unwanted side effects, or that have an exorbitant cost, greatly reduce the patient adherence to the treatment regimen.9–12 Since replacement therapy for hypoparathyroidism (1) requires multiple daily doses of calcium, vitamin D, and Forteo; (2) requires high doses of calcium that can cause kidney stones, constipation, dry mouth, a continuing headache, increased thirst, irritability, loss of appetite, depression, a metallic taste in the mouth, and fatigue; and (3) may include Forteo, which can cost $900–2100 per month; the management plan is often ineffective and could be improved by organ replacement therapy.
Many of the challenges of organ replacement therapy are avoided during parathyroid replacement because of the nature of the parathyroid glands. Complete function of the parathyroid gland is contained within each cell,1,3 the organs have very limited mass, and the cells can be grafted to heterotopic locations.13–15 Also, the parathyroids are an organ system that is affected by a surgical complication rather than a physiological condition or autoimmune disease. The simplicity of the parathyroid gland focuses the treatment challenges on the development of the autologous replacement cells and not on the development of glandular architecture or on protection from autoimmunity.
Our ultimate goal is to isolate autologous human progenitor cells that can be induced to function as parathyroid tissue and that can be grafted as autologous cells. In humans, the thymus develops from the same primordium as the parathyroid glands. 1 Thus, we have investigated thymus as a source of autologous endoderm and parathyroid-like precursor cells. No prior research has been performed to investigate the possible differentiation of thymus into parathyroid cells, although some cells in the thymus normally express parathyroid markers. However, clinical experience shows that parathyroid marker-expressing cells in the thymus are not able to replace parathyroid function in situ in spite of physiologic stimulus due to hypoparathyroidism.
We have previously developed a protocol for nongenetic manipulation of human embryonic stem (hES) cells that induces expression of the parathyroid markers PTH, calcium sensing receptor (CaSR), chemokine receptor type-4 (CXCR4), and chorian specific transcription factor (GCM2).16,17 Our hypothesis in the current work was that a protocol similar to our hESC protocol could be used to induce endogenous precursor cells from thymus to change in similar ways. Our studies show that a subset of human thymus cells can be induced through nongenetic manipulation to express parathyroid markers. The trans-differentiated cells expressed both thymus and parathyroid lineage-specific transcription factors and released PTH in low calcium conditions and elevated calcium concentration in culture suppressed PTH release. Our data suggest that this strategy may be capable of restoring parathyroid function for a patient using in vitro trans-differentiated cells.
Materials and Methods
Thymus cell isolation
Discarded thymus from pediatric cardiac operations was obtained from the operating room and immediately dissociated using the procedure by Mouiseddine et al. 18 While we have human studies approval to also use adult thymus in these studies, pediatric thymus is more cellular than adult, easier to obtain since it is discarded tissue, and does not have to go through pathology evaluation before use, which yields less fresh tissue. Briefly, T cells are released by pressure with the back of a syringe from the thymus tissue, which is covered in HBSS (Hank's balanced salt solution). Tissue is minced and transferred from the HBSS to a triple enzyme mixture 19 (0.1% collagenase, 0.002% deoxyribonuclease, and 0.01% hyaluranidase). The tissue is dissociated in a gentleMACs dissociator (Miltenyi) using the Spleen 2 program (fraction 1). The tissue is incubated at 37°C with gentle agitation for 15 min to yield a second fraction that Mouseddine has characterized as thymic stromal cells (TSC), enriched for multipotent mesenchymal stem cells. The lightly dissociated tissue is further separated using the gentleMACs Spleen 1 program and passed through a 0.22 mm cell screen to separate the cells from the fibrous tissue; these cells are called the thymic epithelial cell (TEC) fraction. This cell fraction from the thymus dissociation process (TEC) was used for further experiments based on expression of precursor markers of interest.
Differentiation protocols
The TEC from thymus were incubated using the Bingham protocol. Briefly, cells were incubated in RPMI 1640 (Invitrogen) with 5% fetal bovine serum (FBS). Cells were kept in culture with or without cholera toxin to slow fibroblast growth and with Activin A (100 ng/mL; R&D Systems) and Sonic hedgehog (Shh; 100 ng/mL; R&D Systems). Cells were tested for marker expression and PTH secretion at each major time point in the protocol. The protocol was modified in subsequent experiments to optimize marker expression.
The differentiated, attached thymus cells were incubated at various time points in Epilife (Gibco) with additions of pen-strep and EPILIFE defined growth supplement (EDGS, Gibco). No FBS was used in this assay. Commercial calcium chloride (Invitrogen) was used for calcium suppression testing. Medium changes and sampling occurred at 24 h time points or took place every 90 min.
Polymerase chain reaction
Total RNA was isolated using TRIzol reagent. cDNA synthesis was performed using the ReactionReady First Strand cDNA synthesis kit (SuperArray). Polymerase chain reactions (PCRs) were performed using primer sets previously published that span exon splicing sites.16,17 GAPD internal standards were used in each reaction. The PCR protocol was one cycle at 95°C for 15 min, and then 30 cycles of 15 s at 95°C, 30 s at 55°C, and 30 s at 72°C with a final extension of 7 min at 72°C.
Enzyme-linked immunosorbent assay
At each major time point of the differentiation protocol, conditioned medium was collected and stored at −80°C until use. The medium was used in a commercial intact PTH ELISA kit (Calbiotech). Cell number was not obtained as the conditioned medium was taken from cells used in the next step of the differentiation process. All samples were tested in triplicate.
Immunofluorescence
Thymus and parathyroid tissue were both dissociated using the protocol described above. The red blood cells were lysed using Ack Lysing Buffer (Lonza) according to the protocol. Cells were also harvested from tissue culture plastic using TryPLE Select (Gibco). Cells were adhered to slides using a Cytospin 4 (Thermo Shandon). Cells were fixed with 2% paraformaldehyde for 15 min at 22°C. For some experiments, CaSR antibody (Catalog #SC-47741, Santa Cruz Biotech) was applied for 1 h at 22°C. Cells were permeabilized in 0.1% Triton X-100 for 6 min at 22°C. Cells were stained with FoxN1 antibody (Catalog #SAB 2500416, Sigma Aldrich) and/or with GCM2 antibody (custom antibody, Primm) for 1 h at 22°C. Coverslips were mounted using ProLong Gold antifade reagent (Invitrogen) containing 4′,6-diamidino-2-phenylindole (DAPI). Stains were observed in a fluorescent microscope.
Results
Complete parathyroid gland differentiation is marked by the specific expression of GCM2. Parathyroid cells are the sole cells in the body that concomitantly produce PTH and express the CaSR as part of their physiologic function to control PTH production and serum calcium levels. Therefore, GCM2, CaSR, and PTH expression represent the definitive markers of the parathyroid cell phenotype to determine the trans-differentiated state of these cells.
The thymus is often discarded during cardiac or neck operations. To determine if human thymus would be a good source of cells for our protocol, we isolated separate populations of thymus cells, including TSC and TEC, according to the protocol by Mouiseddine et al. 18 Reverse transcription (RT)–PCR showed that all of the fractions had low levels of both early endoderm and parathyroid markers, but the TEC fraction had slightly increased expression of markers (Fig. 1). This fraction also contained the most cells. To this end, all of the cells from the TEC fraction were treated with our protocol developed for differentiation of hES cells (Bingham protocol).16,17
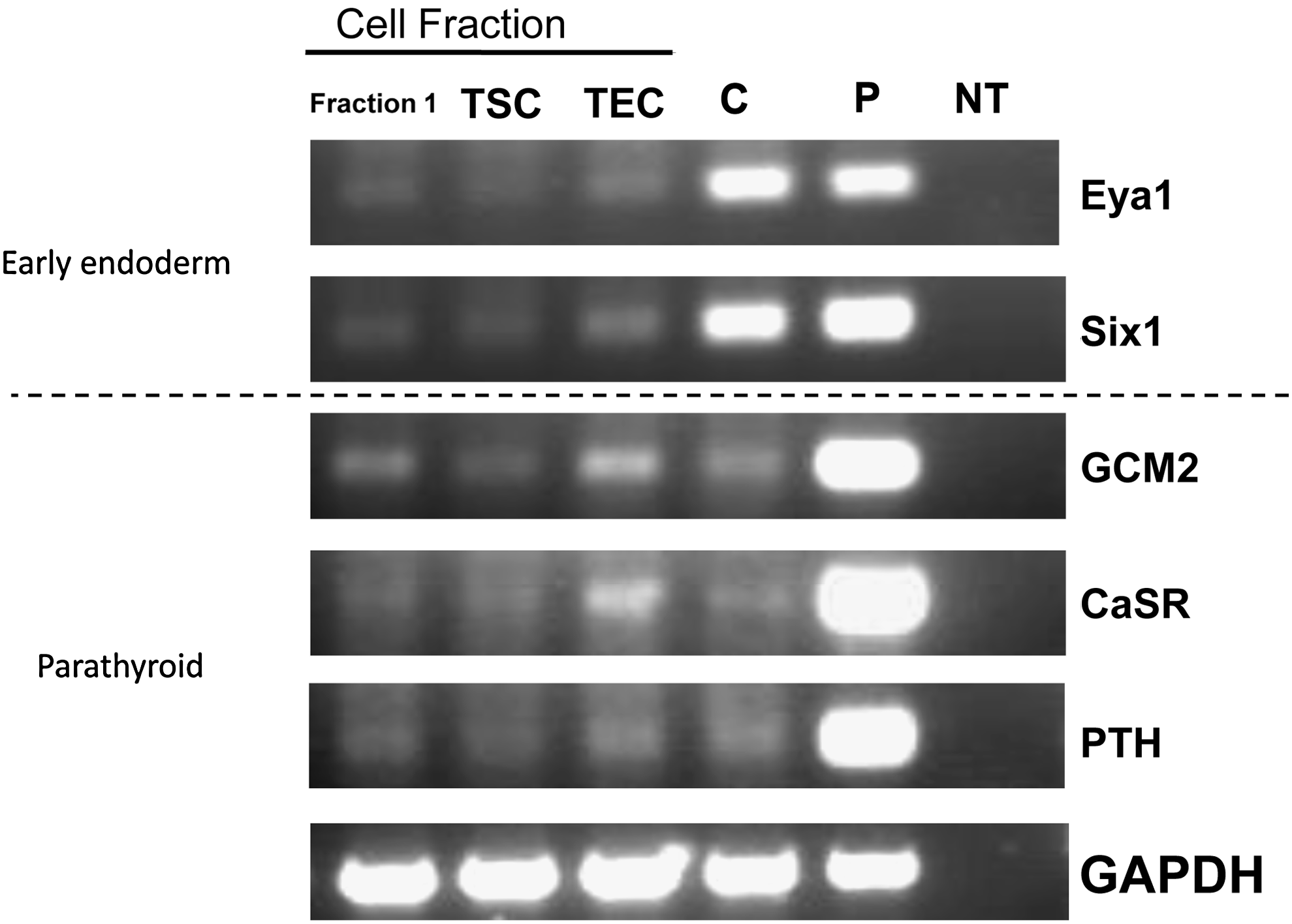
Thymus cells express early endoderm and/or parathyroid markers. Human thymus was dissociated in a series of steps according to Mouiseddine et al. 18 Cells were collected at each fractionation and subjected to reverse transcription–polymerase chain reaction for the early endoderm markers Eya1 and Six 1 and the parathyroid markers GCM2, CaSR, and parathyroid hormone (PTH). Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control. C, commercially available thymus RNA; P, parathyroid RNA; NT, no RNA template used in reaction. Thymic stromal cells (TSC) were released in second fraction and thymic epithelial cells (TEC) are released at the end of the procedure. TEC cells were used in further studies as the expression of appropriate markers was the highest.
TEC that had been in the Bingham protocol for 6 or 10 days with varying amounts of Activin A (bracketing the amount we used in previous studies16,17) were assessed by RT-PCR to determine the expression of early endoderm and parathyroid markers (Fig. 2). Cells in all three concentrations and on both days contained higher amounts of all markers compared with untreated thymus.
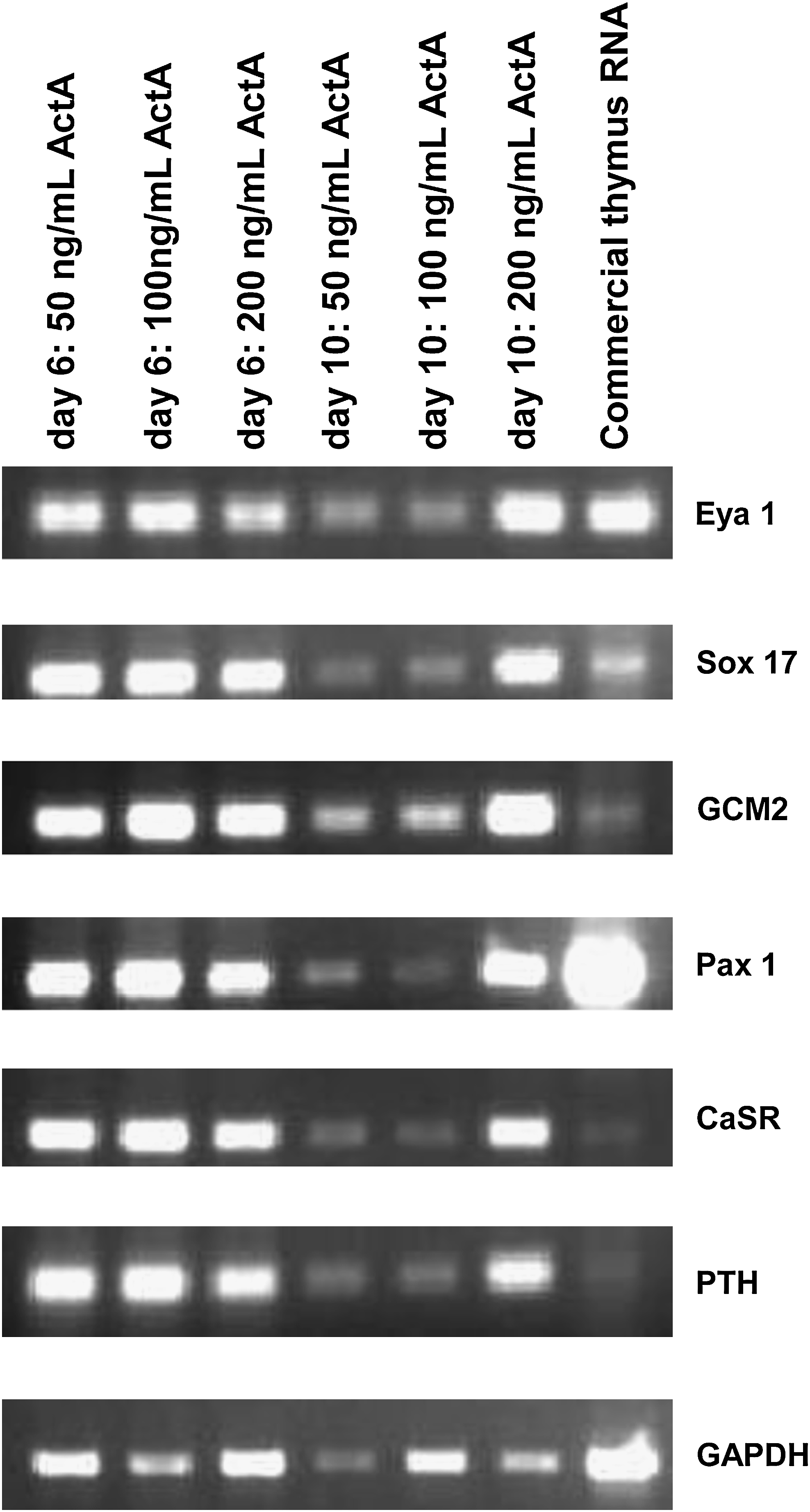
TEC cells could be induced to express higher levels of parathyroid markers. TEC cells (Fig. 1) were treated with the Bingham protocol using a constant dose of Sonic hedgehog (Shh; 100 ng/mL) and varying concentrations of Activin A. Cells were tested at day 6 and 10 for early endoderm and parathyroid marker expression. Cells from the protocol expressed higher levels of parathyroid markers than cells at baseline after isolation and from commercially prepared human thymus RNA (Fig. 1).
Cells in the protocol that replicated throughout adhered to the tissue culture plate during week 6. After adherence, the cells took on an elongated phenotype similar to parathyroid cells in culture (Fig. 3A). Conditioned medium was collected from cells in all three Activin A concentrations at 6 weeks to determine secreted PTH levels. An image of unstimulated thymus cells is presented for comparison (Fig. 3B). Note the structures that resemble thymic corpuscles present in the unstimulated cells. These structures were not observed in the cells that underwent the Bingham protocol. Static levels of PTH secretion from cells that had been through the protocol were modest, similar to those observed with hES cells that had been in the protocol16,17 (data not shown). Calcium-regulated PTH secretion was demonstrated when calcium was withdrawn for 24 h from the cells that had been in the protocol for at least 6 weeks. There was a robust calcium-regulated PTH response after treatment with 50 ng/mL Activin A, half the amount used to differentiate the hES cell16,17 (Fig. 3C). Based on these data, 50 ng/mL Activin A was used in the remainder of experiments. TEC were also kept in culture without the addition of Activin A or Shh as controls but did not secrete PTH under any conditions (data not shown).
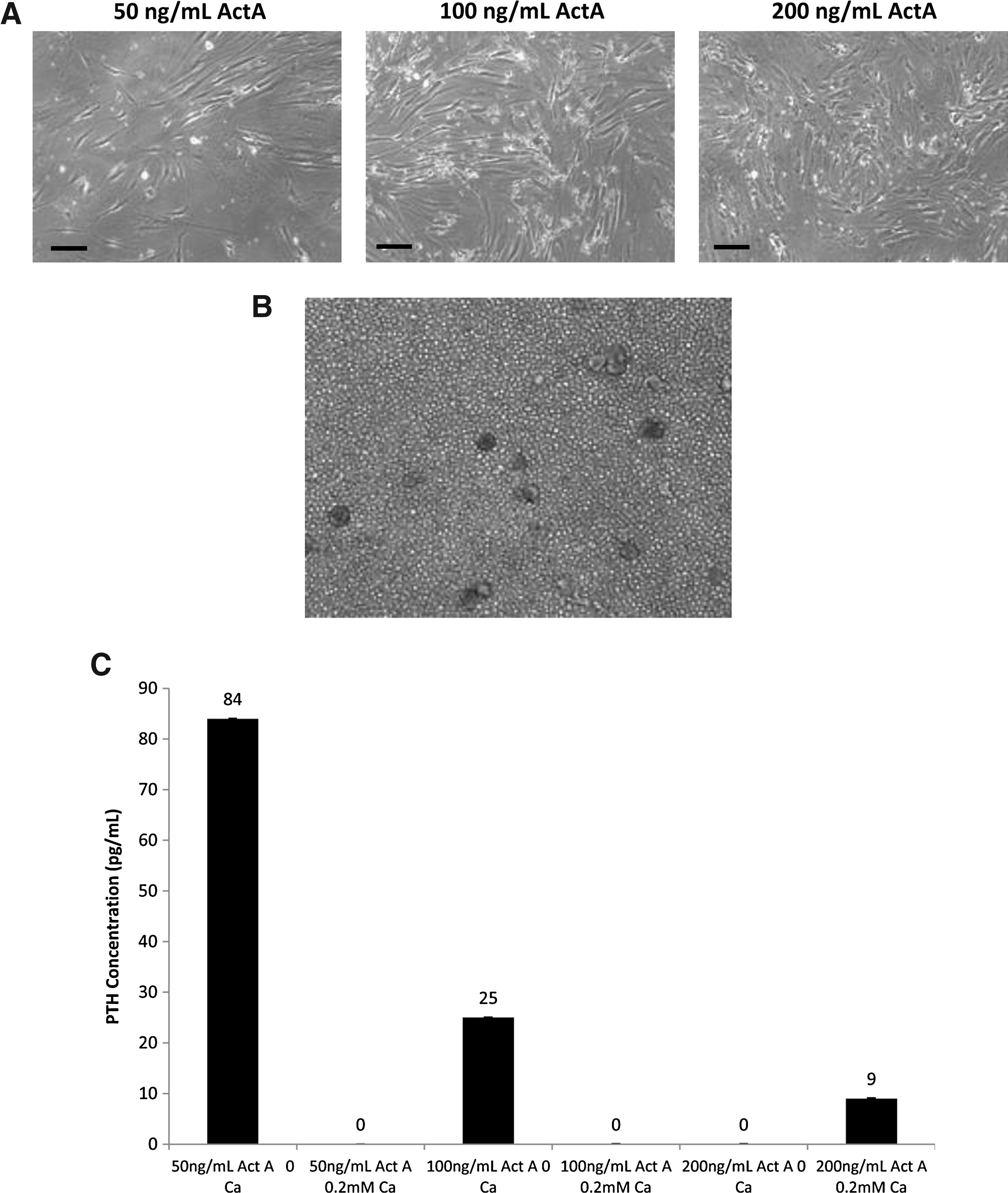
TEC cells from the Bingham protocol secreted PTH in a calcium-dependent manner.
Calcium-regulated PTH secretion was evaluated for timing and durability. TEC were placed in the Bingham protocol with Activin A and Shh. Cells were allowed to grow for 10 days, 3 weeks, and 10 weeks. At each time point, calcium concentrations were changed on identical plates, and the conditioned medium was collected for determination of secreted PTH 24 h later. TEC in the Bingham protocol for up to 3 weeks did not secrete PTH (data not shown); however, the cells had not yet adhered to the tissue culture plates. At 10 weeks, adherent cells secreted large amounts of PTH in response to calcium withdrawal from the culture (Fig. 4A). Levels typically reached 60–84 pg/mL PTH after withdrawal of calcium from the medium. This is slightly elevated from normal for both serum PTH levels and for parathyroid cells in culture that have calcium withdrawn (10–65 pg/mL).20,21 Calcium concentrations between 0 and 0.1 mM do not produce a change in PTH secretion. About 1 to 0.2 mM calcium regularly decreased the secreted PTH levels. Changes of calcium concentrations over 90 min, instead of 24 h, produced secreted PTH only at the 0 mM concentration (Fig. 4B).

PTH release is robust and responsive to calcium withdrawal after cells in the Bingham protocol become adherent.
Using 50 ng/mL Activin A, we determined the optimal concentration of Shh by bracketing the concentration we used previously. 11 After 10 days in the culture protocol, the cells did not secrete any PTH, but by 4 weeks in culture, the cells secreted PTH (Fig. 4A). At 8 weeks, when the cells had fully adhered, the cells secreted PTH in response to calcium withdrawal after growth in 100 ng/mL Shh (Fig. 4C).
Data in the literature 22 suggest that parathyroid cells can be cultured for long periods, but the cells were only useful for experiments for about 1 week. Thus, we evaluated whether growth with Activin A and Shh would allow parathyroid cells to remain functional in vitro. We stained for both CaSR and GCM2 and found that parathyroid cells after 2 weeks lose expression of CaSR protein. Activin A/Shh in the culture medium allowed the parathyroid cells to maintain their parathyroid markers for longer periods (Fig. 5A).
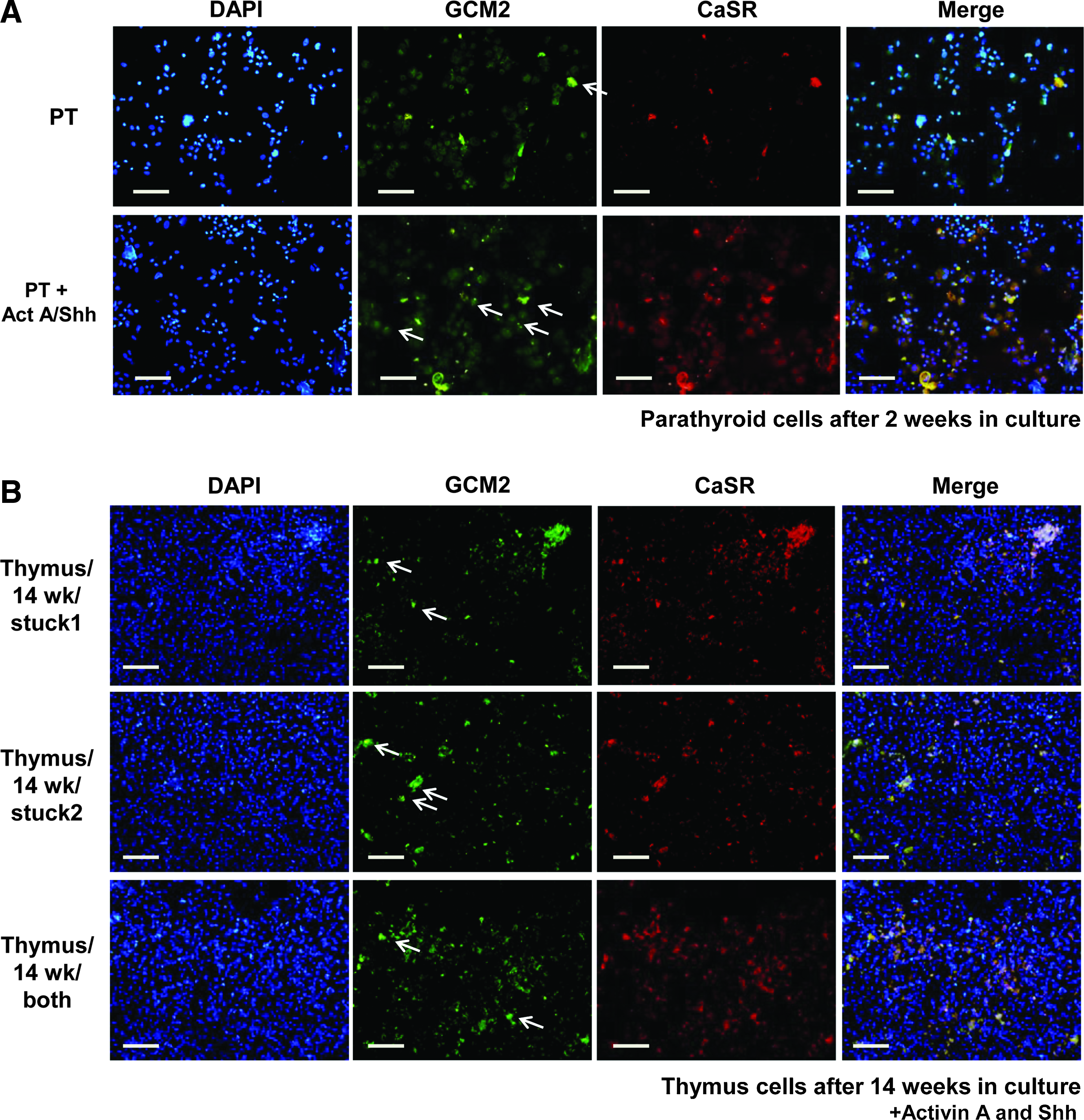
TEC after the Bingham protocol express parathyroid markers and a parathyroid lineage-specific transcription factor. Cells were adhered to slides using a cytospin, fixed, stained for CaSR
Since parathyroid cells under these conditions could grow and express these identifying proteins, we sought to determine if the TEC were trans-differentiating or were expanding a subpopulation of parathyroid-like cells resident in the thymus. We first evaluated TEC that had been in the Bingham protocol for 14 weeks for CaSR and GCM2 expression by immunostaining (Fig. 5B). TEC from the Bingham protocol maintained expression of both CaSR and GCM2. However, evaluation of TEC after the Bingham protocol by staining for lineage-specific transcription factors (FoxN1 for thymus and GCM2 for parathyroid) makes the expansion of thymic resident parathyroid cells unlikely (Fig. 5C). As a control, we stained primary thymus cells for both transcription factors (Fig. 5D), which showed robust staining for FoxN1 but not GCM2. These data show that the majority of the postprotocol TEC are unique in that they express definitive markers of both thymus and parathyroid lineage in the same cell. Note that some of the cells did not stain for expression of any marker and that some cells only stained for expression of one of the markers, indicating that the cell population is not pure.
To determine if our trans-differentiated cells produced tumors in mice, we injected 1×104–1×106 cells subcutaneously into both flanks of 5 RAG2 knock-out mice. We chose RAG2 knock-out mice as the immune-incompetent background for future in vivo experiments. At 3 months after cellular transfer, none of the mice developed tumors. Because our cells were not tagged in anyway, since we do not want any form of genetic manipulation, we were unable to tell if the cells were viable at the end of the experiment. We did replate the cells remaining from the injections, and they did adhere to tissue culture plates. Since the RAG2 knock-out animals have functioning parathyroids, we did not test for PTH secretion by our injected cells.
Discussion
In vitro differentiation of human cells for replacement of organ function is a rapidly developing field. Most research is centered on the addition of developmental genes to redifferentiate cells. Unfortunately, although these genes can drive redifferentiation of the cells, they also facilitate the development of tumors when the redifferentiated cells are placed in mice. Thus, new areas of research center on finding nongenetic methods to drive cells toward redifferentiation.
Many of the challenges of organ replacement therapy are avoided during parathyroid replacement because of the nature of the parathyroid glands: (1) each parathyroid cell contains the complete function of the organ, (2) no architectural arrangement of parathyroid cells is needed to support or enhance the function of the organ, (3) transplantation of a small number of parathyroid cells reconstitutes normal parathyroid function, and (4) patients who have lost this function due to operative complication have no autoimmune reactivity to their parathyroid tissue. Therefore, the parathyroid glands are an ideal system for laboratory-created tissue replacement not only to treat diseases of the parathyroid glands but also as a model for the differentiation of more complex organs. Given the advanced understanding of the molecular markers of parathyroid organogenesis in vivo23–42 and the appropriateness of parathyroid glands for cellular replacement therapy, we have developed methods for recreating parathyroid differentiation using less differentiated cells as a starting point. Beginning with established protocols for generating definitive endoderm from hES cells, 43 we differentiated the hES lines BGO1 and H1 to express pharyngeal endoderm markers by culturing with Activin A and Shh in vitro.16,17 We optimized the culture protocol by serial assessment of the expression of signaling markers in the parathyroid development pathway. Cells that went through the full differentiation protocol expressed the parathyroid markers PTH, CaSR, CXCR4, and GCM2. The differentiated cells also released PTH into the medium. The hES cell lines were used only as a model system for the development of the culture protocols because two main barriers exist to using hES cells for further studies: (1) the potential of these cells and their progeny for alloimmunity and (2) the potential of the cells to form tumors. Building on our observations with the in vitro differentiation of hES cell, we were able to direct the differentiation of TEC into cells with parathyroid function. The process that we detail here results in novel cells that are not precisely the same as native parathyroid cells. Individual cells express markers of both thymus and parathyroid lineages, indicating that they are a new cell type trans-differentiated from thymus precursors, but that also have the unique identifying characteristics of parathyroid cells.
Recently, Bonfanti et al. 44 isolated embryonic rat thymus and redifferentiated them into skin cells by placing them in the presence of rat skin. Although they were able to redifferentiate thymus without genetic manipulation, our studies have key differences. Importantly, the Bonfanti work used embryonic rodent cells on cellular feeder layers. The rodent cells may be a good model for protocol development (rather than the hES cells that we used); however, the use of cellular feeder layers would dramatically complicate eventual clinical applications. In addition, they used environmental factors to create hair follicles, whereas we used developmental factors to trans-differentiate into a functional organ. However, their conclusion that the embryonic rat thymus contains multipotent cells is echoed in our results with developed human thymus.
The weaknesses of our study are (1) that the current experiments are limited to thymus glands from young children having operations for congenital cardiac anomalies and (2) the cultures after differentiation are not likely to contain solely parathyroid-like cells. First, the use of thymus tissue from young children is defined both by convenience and cellularity. With a large number of pediatric cardiac procedures performed in our institution, discarded pediatric thymus is readily available. In addition, the pediatric thymus is very cellular, so each specimen yields a large amount of material to use for experiments. The weakness of this is that the cellular populations in the pediatric thymus may not be the same, or present in the same abundance, in the adult thymus available from the people for whom this strategy would be most applicable. Second, we have not made any significant attempts in this group of studies to isolate the parathyroid-like cells from others in culture. Our fluorescent studies indicate that we have trans-differentiated cells that subsequently make up about 50% of the culture. Isolation of the precise subset will be important to allow specific grafting of the best subset of cells, One strategy is to separate the CaSR-expressing cells to provide as pure a population as possible and then to focus on ways to maximize the PTH production of the differentiated cells.
Taken together, our data show that the postnatal thymus contains cells capable of trans-differentiation into calcium-sensitive PTH-secreting parathyroid-like cells. The presented protocol uses human tissue, recreates a physiologically controlled system (calcium-regulated PTH release), and does not require genetic manipulation of cells. Thus, these data suggest that it will be possible to restore a patient's parathyroid function using in vitro trans-differentiated cells.
Footnotes
Acknowledgments
The authors thank Drs. Jenna Hirsch, Rich Ohye, and Edward Bove from the University of Michigan Health System Department of Surgery for providing thymus from their cardiac surgery patients. We also thank the Department of Surgery for financial support for this work.
Disclosure Statement
There are no potential conflicts of interest associated with any author of this article, and no competing financial interests exist.