
Abstract
Introduction:
Human hair follicle outer root sheath (hORS) cells are known to contain hair follicle stem cells and play an important role in healing large size wounds, and thus can serve as the cell source for skin engineering. This study investigated the effect of low oxygen tension culture on hORS cell proliferation potential and functional maintenance during in vitro expansion.
Materials and methods:
Spared postsurgery scalp tissues were donated by 15 patients aged 20–45 (13 men and 2 women) and were randomly divided into three groups, and isolated hORS cells were combined into three pooled cell samples. They were cultured either in 4% O2 or 21% O2 and were analyzed for cell proliferation, colony forming efficiency (CFE), and their ability in forming engineered skin in vitro.
Results:
The results showed that freshly isolated hORS cells expressed CD200 (22.88±8.76), cytokeratin 15 (CK15) (62.57±4.72), CD29 (22.53±2.49/strong and 29.80±4.09/dim), and CD49f (28.07±15.76/strong and 49.73±5.65/dim). When exposed in 4% O2, hORS cells proliferated significantly faster than the cells in 21% O2 for the first three passages (p<0.05), could better maintain cobblestone morphology, respectively, generate 3.63-folds more and 23.26-folds more cell yields after one and three passages. Additionally, enhanced CFE with significantly higher total and holoclone colony numbers were found in the 4% O2 group than in the 21% O2 group (p<0.05) for the first three passages along with better maintained CK15 expression. Furthermore, hORS cells expanded in 4% O2 could form better epidermal structure of in vitro engineered skin comparing to the skin engineered by the control cells.
Conclusion:
The low oxygen culture method of hORS cells is simple, low cost, less labor intensive, and less biosafety concern, which may potentially be applied in skin engineering and clinical application.
Introduction
Like other engineered tissues, the proper cell source is important for engineering autologous skin grafts to avoid donor-site morbidity. This is particularly true for the treatment of extensive burn injury. In this case, autologous skin donor area is usually difficult to be found for harvesting epidermal cells when the injury area is above 95% and scalp will usually be the available region. To address such a concern, the general approaches would be (1) searching for an alternative donor tissue to provide autologous keratinocytes and (2) developing an optimal culture method that is able to in vitro expand keratinocytes in large quantity without losing cell functions.
Scalp has been used as a donor site for harvesting autologous epidermis or keratinocytes to treat extensive burn injured wounds because of its unique advantage that allows for repeatable harvest of autologous epithelium.4,5 This feature is due to the presence of hair follicle stem cells (HFSCs) that can continuously generate new keratinocytes by asymmetrical division and differentiation.6,7 Actually, the use of hair follicle epithelial cells for generating engineered skin has already been reported, 8 indicating that hair follicle keratinocytes may serve as an alternative cells source for skin engineering, which are generally available even in patients with sever extensive burn injury and able to regenerate themselves after the harvest without donor site morbidity.
In vitro expansion of keratinocytes remains a major challenge because the cultured keratinocytes are prone to become differentiated and senescent, leading to functional loss and apoptosis. 9 Feeder layer cell culture system was developed by Rheinwald and Green to maintain the proliferation potential and functions of in vitro cultured keratinocytes. 10 However, the concern of biosafety has limited its clinical application because of the potential contamination of mouse 3T3 cells that usually serve as the feeder layer cells. 11
In vivo, human cells are exposed to an environment with the oxygen tension much lower than that of in vitro cell culture environment. Therefore, the reduction of oxygen tension in cell culture is considered more physiologically relevant to the in vivo niche of the cultured cells.12,13 In previous reports, low oxygen tension (hypoxia) culture has been applied to the in vitro culture of several kinds of stem cells, including embryonic, mesenchymal, hematopoietic, and neural stem cells, and proved beneficial for cell proliferation and functional maintenance.14–17 However, there is no report yet about the effect of hypoxia on cultured human hair follicle outer root sheath (hORS) cells, and these cells are known to contain epidermal stem cells. 18 The aim of this study was to explore the potential benefit of low oxygen culture for expanding functional hORS cells in vitro.
Materials and methods
Isolation of hORS cells and dermal fibroblasts
Protocols for the handling of human tissue and cells were approved by the Ethics Committee of Shanghai 9th People's Hospital. Total 15 patients were involved in this study including 13 men and 2 women with the age between 20 and 45. Human abandoned occipital scalp tissues were donated by the patients who received hair transplant surgery with informed consent. hORS cells were isolated according to previous reports.19,20 Briefly, after being washed in phosphate buffered saline (PBS), the scalps were cut into 1×0.5 cm pieces, and the subcutaneous fat tissue was removed following by the treatment with 0.1% Dispase II(Roche) in Dulbecco's modified Eagle's medium (DMEM; Gibco) overnight at 4°C to mechanically separate the epidermis and dermis. The samples were examined under a microscope, and only the hair follicles in anagen stage were selected and pulled out with fine forceps, and the dermal papillae were cut off to avoid the contamination of dermal papilla cells. Then, the isolated follicles were incubated with 0.05% trypsin–ethylenediaminetetraacetic acid (EDTA) (Gibco) for 20 min at 37°C to isolate hORS cells. After neutralization with DMEM containing 10% fetal bovine serum (FBS; Hyclone), the hORS cell suspension was filtered through a 70-μm cell strainer (BD Falcon) followed by centrifugation at 800 g for 5 min. The cell pellet was then resuspended in Defined Keratinocyte Serum Free Medium (dK-SFM; Gibco) and the viable cell counts were determined using a hemocytometer after the staining with 0.4% Trypan Blue. To perform the study, hORS cells from every five patients were combined as one pooled cell sample; thus, the cells from total 15 patients were pooled into three cells samples for various experiments with three repeats.21,22
To engineer skin tissue, foreskin specimens were obtained from the children who received circumcision surgery with informed consent to their parents. After being washed in PBS, the specimens were incubated with 0.1% Dispase II in DMEM overnight at 4°C, and then the epidermis was mechanically separated from the dermal part. Afterward, the dermis was cut into small pieces and digested with 0.1% Collagenase NB (SERVA) in DMEM at 37°C for 2 h to isolate dermal fibroblasts. The suspension was filtered through a 70-μm cell strainer and centrifuged at 800 g for 5 min and the cell pellet was resuspended in the DMEM culture medium.
Cell culture and passage
The isolated primary hORS cells were seeded onto six-well plates (BD Falcon) at the density of 2000/cm2 in dK-SFM supplemented with 10 ng/mL hEGF (Peprotech), 10 ng/mL cholera toxin (Enzo), 100 U/mL penicillin–streptomycin (Gibco), and 25 mM HEPES (Gibco). Afterward, the hORS cells were cultured and passaged either in low oxygen tension (experimental group) or in regular oxygen tension (control group). Briefly, the cells of experimental group were cultured in a CO2 incubator (Series CB, Binder) with 4% O2; the control cells were cultured in a CO2 incubator (Forma Science) with 21% O2 and the media were changed every 3 days. When reaching 80% confluence, the hORS cells were passaged with the treatment of 0.05% trypsin-EDTA for 5 min at 37°C to detach the cells. Then, the trypsin was neutralized with DMEM containing 10% FBS. After centrifugation at 800 g for 5 min, the cell pellet was resuspended in dK-SFM medium and seeded onto 6-well plates at the same density of 2000/cm2 for subculture.
For human dermal fibroblasts culture, the isolated cells were seeded onto 10-cm culture dishes in DMEM culture medium containing 10% FBS, L-glutamine (292 mg/mL; Gibco), penicillin (100 U/mL; Gibco), streptomycin (100 mg/mL; Gibco), and ascorbic acid (50 mg/mL; Sigma), and incubated at 37°C in a humidified atmosphere containing 95% air and 5% carbon dioxide. The medium was changed every 2 to 3 days, and the cells were subcultured when they reached 80% confluence.
Generation of cell growth curve and cell cumulative doubling curve
To determine the effect of oxygen tension on cell proliferation, the harvested hORS cells were seeded onto 24-well plates (BD Falcon) at the density of 2000/well and cultured in either low or normal oxygen tension as above described. At 24 h, the cells of both groups were trypsinized and counted with an automatic cell counter (Z2 coulter, Bechman Coulter) as day 1 cell number. Afterward, cells were harvested for counting every 48 h to obtain the data of days 3, 5, 7, 9, and 11 (for passaged cells) or to day 17 (for primary cells). To reduce variation, cell counting was performed in quadruplicated wells and cells of each well were counted three times to obtain the mean cell number and standard deviation.
To generate cumulative cell doubling curve, the numbers of the cells harvested at each passage (averaged cell number per 6-well plate) were divided by their respective seeding numbers to generate the folds of cell expansion (X) of each passage, which was then transformed into the number of cell doublings (n
Calculation of cell doubling times
According to the previous report, 23 the population doubling times during the exponential growth phase were determined by the formula: PD=(t2 – t1)×Lg2/(LgN2 – LgN1), where PD=population doubling times; t2=times at the end of exponential growth phase (days); t1=times at the beginning of exponential growth phase (days); N2=the cell numbers at the end of exponential growth phase; N1=cell numbers at the beginning of exponential growth phase.
Colony-forming efficiency assay
To determine the effect of low oxygen tension on colony forming efficiency (CFE), the primary hORS cells were seeded onto 10-cm dishes at the density of 5000/dish and cultured for 14 days in either low or normal oxygen tension as above described. Additionally, passages 1 to 5 hORS cells cultured in either low or normal oxygen tension, as described in the section of Cell culture and passage, were, respectively, harvested and seeded onto 10-cm dishes at the density of 1000/dish and continued culture, respectively, in low or normal oxygen tension for 14 days. At day 14 time point, all plates were collected and the colonies were fixed in 4% paraformaldehyde (Sigma) in PBS for 10 min at room temperature. After being washed in PBS, the colonies were stained with 1% Rhodamine B (Sigma) in water for 30 min at room temperature, and were photographed after another three washes in water. CFE was determined by the ratio of colony number to initial plating cell number and expressed as a percentage as previously reported. 24 Results were presented as means±standard deviation. The colony-forming unit containing 50 or more cells was scored as holoclone as previously reported.25–27 The final CFE was determined by averaging the numbers of quadruplicated dishes.
Flow cytometry analysis of Cytokeratin-15 and surface antigens
The isolated primary hORS cells or cultured hORS cells harvested with trypsinization were filtered through a 40-μm cell strainer and washed in PBS after centrifugation at 400 g for 5 min. Briefly, the collected hORS cells were incubated with the antibodies of PE conjugated mouse anti-human CD200 (BD Pharmingen; 1:200 in PBS) or PE conjugated anti-human α6-integrin (CD49f, eBioscience; 1:200 in PBS) in dark at 4°C for 30 min. After two washes in PBS, the hORS cells were resuspended in PBS containing 1% FBS and ready for flow cytometry analysis (FCM) analysis. For β1-integrin (CD29) analysis, hORS cells were incubated with mouse anti-CD29 monoclonal antibody (Abcam; 1:200 in PBS) at 4°C for 30 min followed by two washes in PBS, the cells were then incubated with goat anti-mouse IgG Alexa Fluor 488 (Invitrogen; 1:1000 in PBS) in dark at 4°C for 30 min and washed twice in PBS. For cytokeratin (CK) 15 expression analysis, the cells were first fixed in 4% paraformaldehyde at 4°C for 15 min, washed in 0.2% Tween-20 (Amresco) in PBS, and then permeabilized in PBS containing 0.2% Tween-20 in dark at 4°C for 15 min. After centrifugation, the cells were blocked in PBS containing 5% FBS at 4°C for 30 min, and then incubated with rabbit anti-CK15 monoclonal antibody (Abcam; 1:200 in PBS) at 4°C for 30 min. After two washes in PBS containing 0.2% Tween-20 again, the cells were incubated with goat anti-rabbit IgG-PE (Santa Cruz; 1:1000 in PBS) in dark at 4°C for another 30 min. After the final wash in PBS, all cells were resuspended in PBS containing 1% FBS and subjected to FCM analysis with Epics ALTRA (Beckman Coulter). The mean percentage of positive cells was derived from the averaged positive cell percentage of three experiments. For each test, a respective isotype antibody served as a control.
Skin engineering with hORS cells expanded in different oxygen tensions
The construction of tissue-engineered skin was performed as previously described. 28 To prepare the scaffold, rat tail collagen I stock solution (5 mg/mL; Shengyou) was diluted to 1.5 mg/mL according to the manufacturer's instruction. For example, to obtain 1 mL of 1.5 mg/mL collagen I solution, 300 μL of 5 mg/mL collagen I stock solution was added into 18 μL of 0.1N sodium hydroxide (Sigma) to neutralize the acetic acid in the collagen. After vibration, 35 μL of 10×PBS (Gibco) and 647 μL of DMEM containing 10% FBS or fibroblasts suspension were separately mixed into the neutralized collagen. The cell free collagen dilution or the dilution mixed with fibroblasts was ready to use. Briefly, 1 mL of the collagen dilution was added to the six-well millicell hanging cell culture insert (Millipore) and polymerized at room temperature for 20 min. Afterward, 3 mL of collagen dilution containing 25,000/mL foreskin fibroblasts were added on the top of acellular collagen layer and then allowed the gel to polymerize at 37°C for 2 h, this construct was then cultured in DMEM containing 10% FBS for a week to form a dermal equivalent. After 1 week, passage 1 hORS cells (total 10,000) derived from both experimental and control groups were, respectively, seeded on the top of each of dermal equivalents and incubated for 2 h at 37°C to allow for cell attachment. Afterward, the complex skin equivalents were cultured in submerge fashion in dK-SFM supplemented with 1.5 mM CaCl2 (Sigma), 0.3% FBS for 1 week. Subsequently, the hORS cells were allowed for air exposure by switching the skin construct to air–liquid interface and cultured in dK-SFM containing 1.5 mM CaCl2 and 2% FBS for 1 week, and then the FBS was reduced to 1% for another week of culture. Afterward, the engineered skin equivalents were harvested and subjected to histological examination. The procedures were repeated three times with three pooled cell samples.
Hematoxylin and eosin staining
The harvested skin equivalents were fixed in 4% paraformaldehyde at 37°C for 2 h, and the specimens were then embedded in O.C.T. compound (Sakura Finetek), frozen sectioned to the thickness of 8 μm followed by hematoxylin and eosin staining.
Statistical analysis
The Mann–Whitney U test (single tail) was applied for statistical analysis. Data were represented as means±standard deviation. A p-value of less than 0.05 was considered statistically significant.
Results
Characterization of isolated human outer root sheath cells
To characterize isolated hORS cells, the expression of human HFSC related molecules were examined with FCM analysis. As shown in Figure 1, among the isolated hORS cells, 62.57% of them expressed CK15. Additionally, 22.88% expressed CD200, which is a relatively specific marker for human HFSCs. Differently, there were apparently two subpopulations of the cells that expressed CD29 and CD49f. As shown in Figure 1, 22.53% cells had strong expression of β1-integrin (CD29), whereas 29.80% cells had dim expression of CD29. The results also revealed that 28.07% cells expressed α6-integrin (CD49f) strongly, whereas 49.73% cells had dim expression of CD49f. These results indicated that primary isolated hORS cells contained certain amounts of HFSCs.
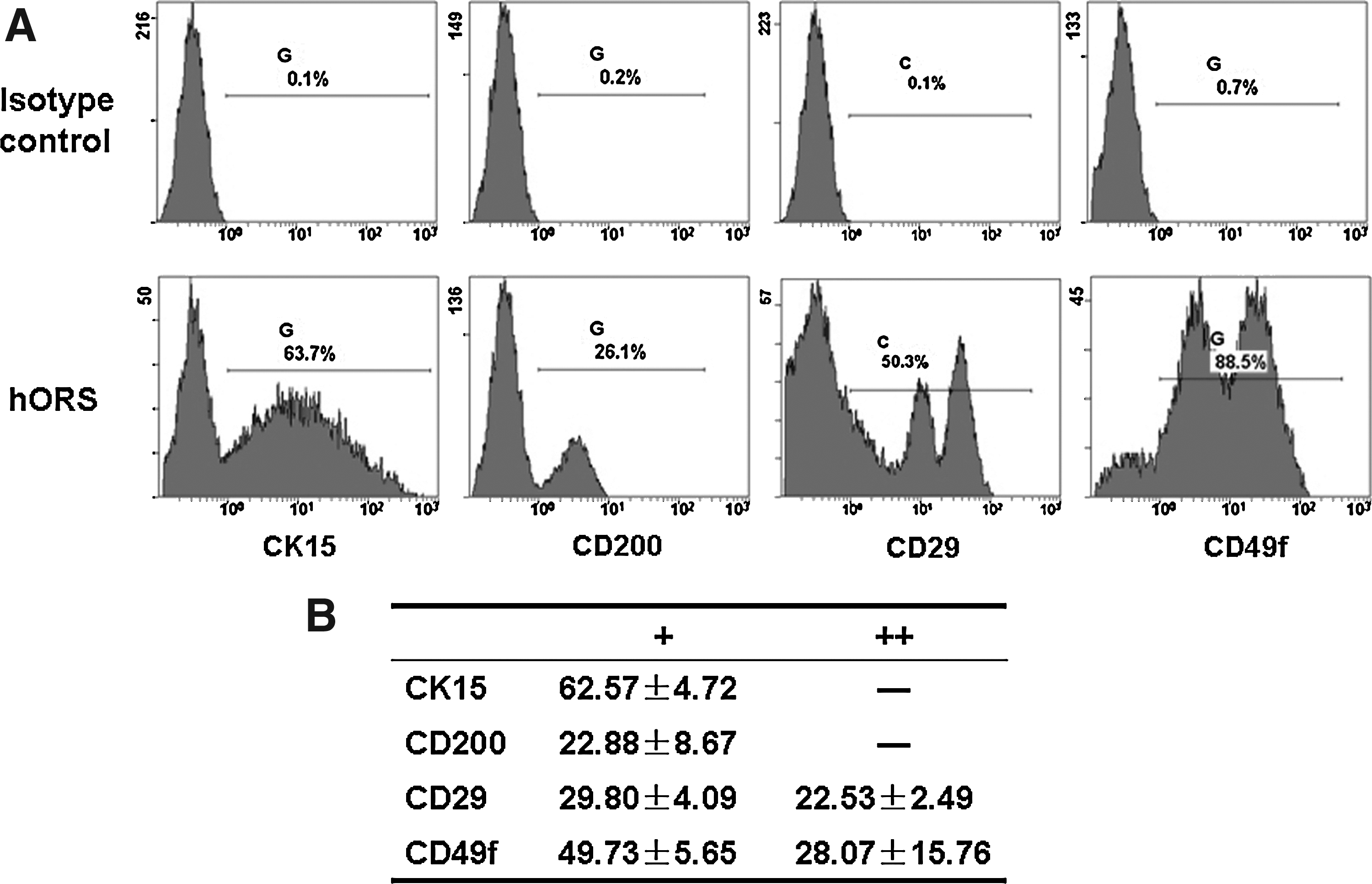
Characterization of freshly isolated hORS cells by flow cytometry.
Low oxygen tension culture promoted hORS cell proliferation
The results of this study revealed an obvious effect of low oxygen tension on cell behavior. At primary culture, hORS cells exhibited typical keratinocyte morphology of cobblestone cell shape. As expected, with cell passage in normal oxygen tension, hORS cells gradually lost this character and became larger in their size as shown in Figure 2. This morphological change became particularly obvious after passage 3, suggesting that cultured hORS became differentiated and were prone to senescent when they grew in vitro without the support of feeder layer cells. Interestingly, when exposed in 4% O2, the cultured hORS cells exhibited an apparently compact cell distribution and smaller cell size at primary culture as opposed to the cells grown in 21% O2 (Fig. 2). At later passages, both group cells became significantly aged with an obvious spread and irregular cell morphology, suggesting that low oxygen tension has a transient beneficial effect for maintaining the function of cultured hORS cells.
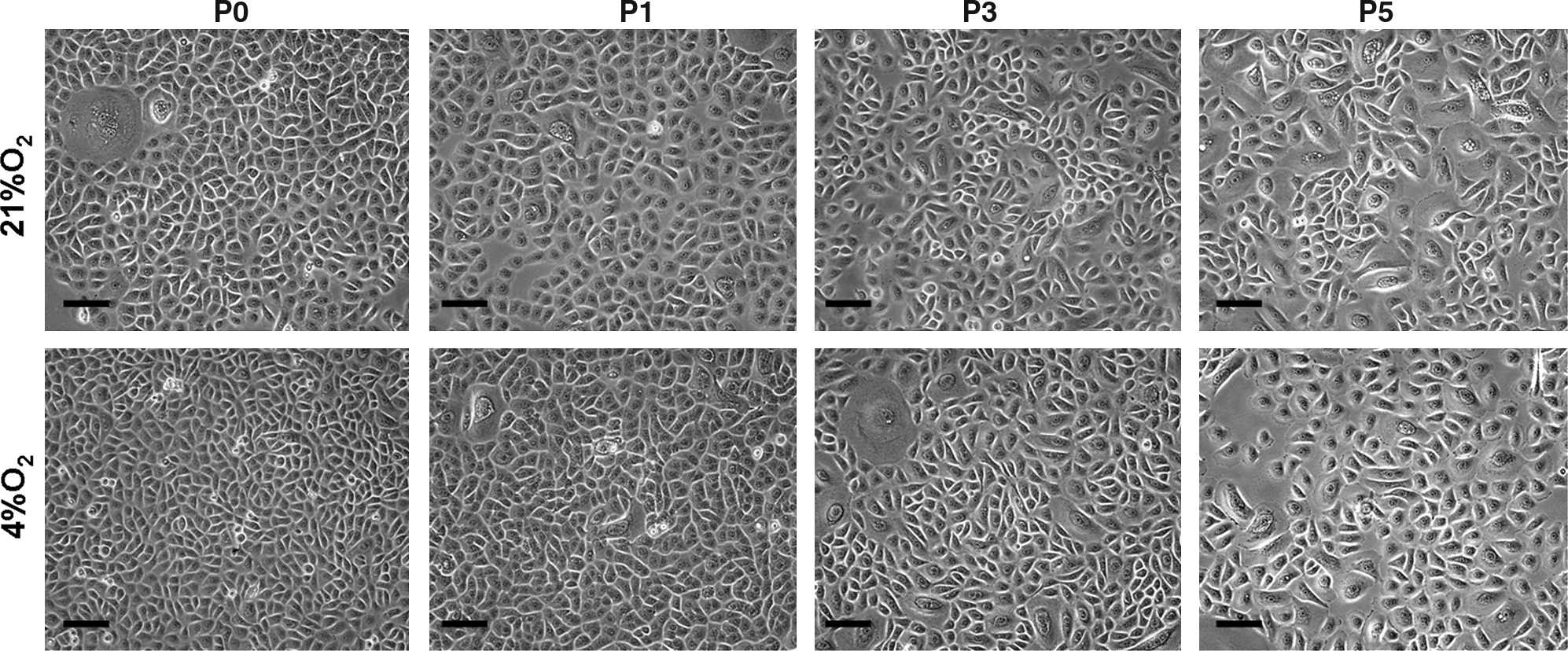
The morphological difference of hORS cells cultured in different oxygen tensions. Cells cultured in 4% O2 reveal smaller cell sizes and more compacted distribution than the cells in 21% O2. Scale bar=20 μm. P0, P1, P3, and P5 respectively represent passages 0, 1, 3, and 5.
To further relate the finding of improved cell morphology to the improved cell function, cell proliferation potential was evaluated by the comparison of cell growth curves of two group cells. The results revealed an apparent benefit of low oxygen tension culture for enhancing cell proliferation potential. As illustrated in Figure 3, hORS cells grew much faster in 4% O2 than in 21% O2 during the exponential growth phase, and the fast growth rate resulted in more cells being expanded during this time period. To determine the difference in growth rate between two groups, quantitative analysis of cell doubling times were performed. As revealed in Table 1, low oxygen tension culture could significantly shortened the cell doubling times comparing to that of normal oxygen tension in the first three passages (p<0.05), but not in passages 4 and 5 (p>0.05).

The growth curve of hORS cells cultured in different oxygen tensions at primary culture
The population doubling times (days) in exponential growth phase were calculated with the formula: PD=(t2 – t1)×Lg2/(LgN2 – LgN1). 23
Although there was a transient effect of functional enhancement by culturing the hORS cells in low oxygen tension, the difference in cumulative cell doublings was obvious. As shown in Figure 4 and Supplementary Table S1 (Supplementary Data are available online at

The difference in the cell doubling numbers between the hORS cells in 4% O2 and 21% O2. The difference is determined by subtracting the numbers of 21% O2 group from those of 4% O2 group. The symbol “-” and the numbers represent how much more cell doublings can be achieved by proliferating cells in 4% O2 than in 21% O2.
Low oxygen tension enhanced CFE of cultured hORS cells
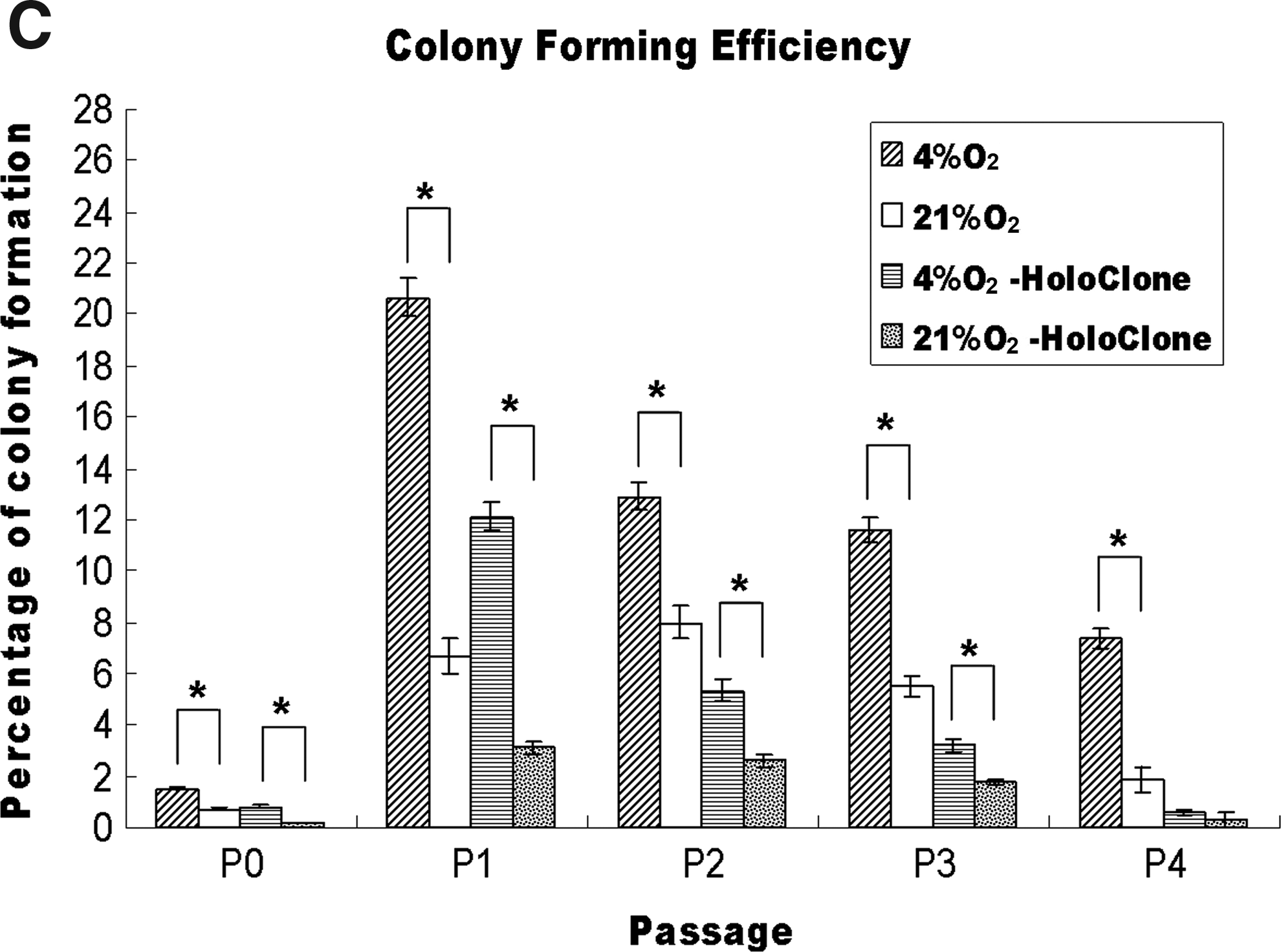
The capacity of colony formation was considered as one of the characters of epidermal stem cells, 29 therefore the CFE assay was used to examine the effect of oxygen tension on the function of cultured hORS cells. As showed in Figure 5A, from primary cells to passage 4, cells cultured in 4% O2 formed more cell colonies than the cells in 21% O2, but both group cells lost their ability to form cell colonies at passage 5 and later passages.


According to literature, 27 keratinocytes cultured on feeder layer cells could form three types of cell colony: holoclone (high proliferative potentials), meroclone (partial proliferative potentials), and paraclone (terminal differentiation). Such findings have not been reported for epidermal cells grown on nonfeeder layer culture system. In this study, similar colony types were observed in hORS cells cultured on regular dish, and they were defined as the following categories: Holoclone, characterized by its large colony size and compactly arranged small size cells (Fig. 5Ba); Meroclone, containing both small and large cells that were arranged in a scattering fashion (Fig. 5Bb); Paraclone, characterized with its small colony size and containing some large size cells (Fig. 5Bc). It was, however, found that the holocolones were much easier to be identified and separated from the other two types of colonies. Unlike feeder layer culture system, the meroclones and the paracolones were relatively difficult to be distinguished from each other; therefore, holoclone and total colonies were used as the parameters for quantitative analysis in this study.
As shown in Figure 5C, it was observed that the colony numbers were relatively low in primary cultured cells (below 2% of total seeded cells), but sharply increased after cell passage and peaked in the first passage, which was about 20% of total seeded cells in 4% O2, but was only about 6% for the cells in 21% O2. The passages 2 and 3 cells remained able to form relatively high numbers of cell colonies with 13% and 11%, respectively, of total seeded cells in 4% O2 as opposed to about 8% and 5.5% respectively for the cells in 21% O2. More importantly, the holoclone percentages of passages 1, 2, and 3 reached 12, 5.8, and 3.8 percentages, respectively, for cells in 4% O2, which were significantly higher than their counterparts cultured in 21% O2. Statistical analysis showed a significant difference between two groups of cells in total cell colony numbers (p<0.05) and in holoclone colony numbers (p<0.05) from primary culture to passage 3. However, at passage 4, the significant difference was only found in total cell colony numbers (p<0.05) but not in holoclone colony numbers (p>0.05), because they were barely detectable (Fig. 5A). These findings suggest that low oxygen tension culture may help the cultured hORS cells to better maintain their functions during their early passages of in vitro expansion.
Low oxygen tension enhanced CK15 expression
FCM analysis revealed that the expression rates of CK15 were significantly higher in 4% O2 group than in 21% O2 group with the percentage of 60.47%±12.34% versus 38.97%±3.42% at passage 1 (p<0.05), 48.9%±4.91% versus 32.33%±4.96% at passage 2 (p<0.05), and 35.67%±2.9% versus 28.83%±3.76% at passage 3 (p<0.05) (Fig. 6), indicating that low oxygen tension might be beneficial for contained HFSCs to better maintain their stemness during in vitro culture.
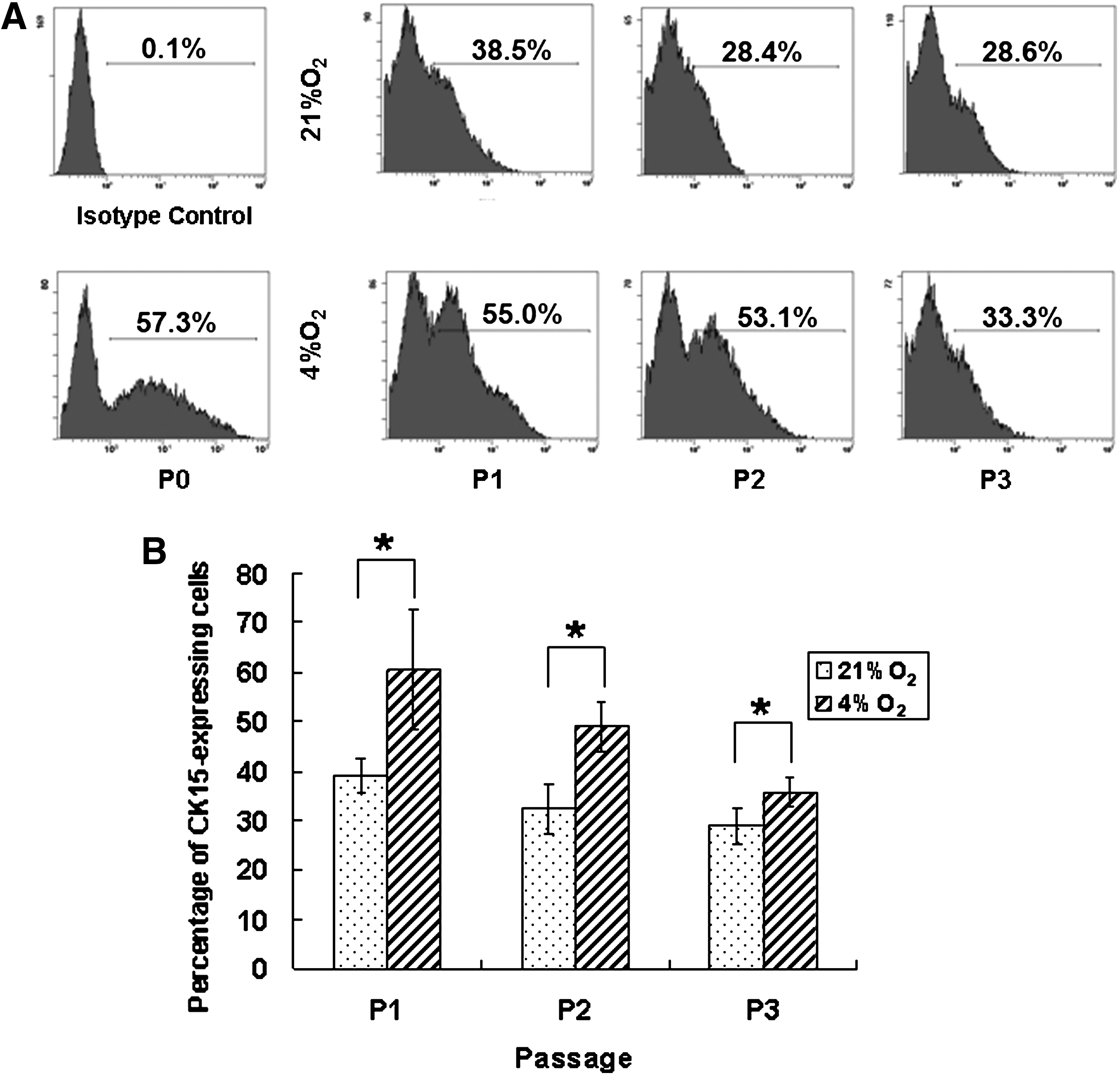
The flow cytometry analysis of CK15 expression in hORS cells expanded in vitro in different oxygen tensions.
hORS cells expanded in low oxygen tension generated better engineered skin
To further evaluate the function of expanded hORS cells, in vitro skin engineering was used as a model. As showed in Figure 7, a full-thickness skin containing both epidermal and dermal layers was able to be generated by collagen scaffold, dermal fibroblasts, and cultured hORS cells. Interestingly, the cells expanded in 4% O2 formed a thicker epidermis with a better tissue structure including multiple keratinocyte layers and the formation of an apparent stratum corneum, indicating that these expanded cells could form a relatively mature epidermis. In contrast, epidermal structure formed by the cells expanded in 21% O2 was relatively poorly developed and less mature. These findings indicate that low oxygen tension culture could not only enhance the cell proliferation potential but also improve their capability of forming epidermal structure of engineered skin.

Hematoxylin and eosin staining of the skin engineered with the hORS cells in vitro expanded in different oxygen tensions. A thicker epidermis with multiple cell layers and an obvious stratum corneum is formed by the cells expanded in 4% O2 (right panels). In contrast, a thinner epidermis with less cell layers and less developed stratum corneum is formed by the cells cultured in 21% O2 (left panels). Bottom panels represent the magnified pictures of top panels, and Scale bar=50 μm. Color images available online at
Discussion
Outer root sheath (ORS) is the external layer of the hair follicle that is contiguous with the basal layer of the interfollicular epidermis, and human ORS cells can also serve the function of keratinocytes for wound closure. 30 Because human HFSCs are located at the specific area of ORS, which is limited to the bulge region where arrector pili muscle attaches; thus, isolated hORS cells also contain HFSCs. 25 HFSCs are usually characterized with related markers of CK15, CD200, CD29, and CD49f,25,31–33 and the finding of these marker expression from freshly isolated hORS cells (Fig. 1) also provided the supporting evidence. As reported, ORS cells have already been applied to skin engineering 34 because of its availability and the functions of self-renewal and differentiation of contained HFSCs, which allow for engineering a skin with better functions.35,36
Like epidermal cells, in vitro expansion of hORS cells with maintained functions remains difficult because these cells easily become aged and senescent, and this is likely due to the deprivation of their in vivo niche factors. Mimicking in vivo niche environment is considered important for preventing cell aging process during in vitro expansion. For example, the feeder layer cell culture system was developed for the expansion of epidermal stem cells 10 or HFSCs 37 in order to mimic the epithelial-mesenchymal interaction signaling for better preservation of the cell function.38,39
It is generally recognized that the bulge area constitutes the important niche for HFSCs to maintain their stemness such as self-renewal function. There might be many factors that constitute such a special niche including particular extracellular matrix, growth factors and cell–cell interactions. 13 Although different tissues may have their own niche factors to support specific stem cells, it was found that one factor is common for virtually all stem cell niches, that is, low oxygen tension. It has been reported that hypoxia could promote the proliferation and maintain the undifferentiated state of several kinds of stem cells, including embryonic, mesenchymal, hematopoietic, neural, and adipose-derived stem cells.14–17 In fact, 2%–9% oxygen tension is quite common in vivo in various tissues and organs and is known as physiological normoxia 13 ; thus, several studies have shown that 5% O2 could enhance the generation of induced pluripotent stem cells, 40 and 3%–5% O2 was considered as an appreciable oxygen tension in stem cell culture. 12
This study also showed that such beneficial effect could be achieved for in vitro expanded hORS cells, including significantly enhanced proliferation potential and better preserved functions. As shown in Figure 4, simply culturing the cells in 4% O2, 1.86 more cell doublings or 3.63-folds more cells could be obtained after 1 passage and 4.54 more cell doublings or 23.26-folds more cells could be expanded after 3 passages as opposed to normal oxygen tension culture. This finding is important for future practical application, because much less hair follicles are needed to generate enough cells for autologous skin engineering when comparing to the routine cell expansion procedure. Additionally, this method is simple, low cost, and less labor consuming and thus is easy to perform. Moreover, because there is no need for adding complicated ingredients into the medium and no concern for contamination of feeder layer cells, the hORS cells expanded in low oxygen tension may become a biosafe cell source for skin engineering and application. Furthermore, low oxygen-expanded cells formed better epithelium structure (Fig. 7), suggesting the beneficial effect for skin engineering. Importantly, further characterization of their functions in vivo such as wound repair and barrier function will be an important direction in future studies.
As this study primarily focused on developing a practical hORS cell culture method, detailed mechanism study in depth would be beneficial for further optimizing this method. Nevertheless, the enhanced CFE (Fig. 5) and better maintained expression of CK15 (Fig. 6) may suggest that low oxygen is likely to better maintain the stemness of contained HFSCs, which also agrees with the similar findings of other types of stem cells.14–17 Although not shown, CD200 expression was not detected by FCM after cell passages in both groups. By contrast, the expression levels of CD29 and CD49f increased to above 90% with cell passage (data not shown), and this phenomenon is likely due to the artifact caused by cell culture, which has been observed in other adhesion molecule expression as well, for example, CD105. 41
In this study, a transient beneficial effect was observed only for the first 3 passages, suggesting that oxygen tension simulation alone may not be sufficient for mimicking natural hORS in vivo environment. Additionally, frequent exposure under normal oxygen tension during medium change may also lead to this transient effect. In the future, combined effect of hypoxia with other niche factors should be investigated for enhancing the functions of expanded hORS cells. Additionally, strict control of oxygen environment for cell culture procedure as well as for the culture of engineered skin, such as a closed hypoxia worktable, will be a more appropriate approach for future investigation.
Footnotes
Acknowledgments
This study was supported by National Natural Science Foundation (30872694) and National “863” (2006AA02A127) and “973” (2005CB522703) Project Foundation.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.