
Abstract
Tissue engineering of clinically applicable dermo-epidermal skin substitutes is crucially dependent on the three-dimensional extracellular matrix, supporting the biological function of epidermal and dermal cells. This matrix essentially determines the mechanical stability of these substitutes to allow for safe and convenient surgical handling. Collagen type I hydrogels yield excellent biological functionality, but their mechanical weakness and their tendency to contract and degrade does not allow producing clinically applicable transplants of larger sizes. We show here that plastically compressed collagen type I hydrogels can be produced in clinically relevant sizes (7×7 cm), and can be safely and conveniently handled by the surgeon. Most importantly, these dermo-epidermal skin substitutes mature into a near normal skin that can successfully reconstitute full-thickness skin defects in an animal model.
Introduction
Almost perfect closure of full-thickness wounds can be achieved by the transplantation of healthy, autologous, full thickness skin. However, only relatively small areas (<2% of the body surface) can be covered by full-thickness skin grafts, as the harvesting of these creates massive trauma at the donor sites. Allogenic full-thickness skin (e.g., donor skin) can only be used as temporary wound coverage, as it will be immunologically rejected with time. 4 A third option for the surgeon are autologous split thickness skin grafts, which consist of the complete epidermis and a thin dermal portion. The corresponding procedure is the current “gold standard” that treats third-degree skin defects. Since there is almost always a complete recovery of the donor sites, this method represents a reasonable solution and is, therefore, widely used. 5 However, a drawback of this method is that, particularly for skin defects larger than 60% bovine serum albumin (BSA), the availability of healthy split thickness skin is limiting.3,5
Superior healing and good long-term function of split thickness skin are largely dependent on a functional dermis. Thus, it is absolutely crucial in skin reconstitution to restore a high-quality dermal compartment. Acellular dermal templates such as IntegraDRT®, used in combination with split-thickness skin transplants, have brought about certain improvements regarding long-term functional and cosmetic results, but this treatment requires a two-step, staged procedure.6–8
We describe here the development of a clinically applicable, autologous full-thickness skin substitute, explicitly including a functional dermal compartment. It is a centrally positive effect of the dermal fibroblasts that these essentially support the deposition of a basement membrane and the proliferation, differentiation, and stratification of epidermal keratinocytes.9–11 This is most likely due to the permanent production of a set of biologically active growth factors that can act in physiological concentrations at their appropriate locations.
Our previous work has provided evidence for the efficiency of collagen type I hydrogels in supporting the generation of organotypic epithelial structures and dermo-epidermal skin substitutes.12–16 The drawback of conventional collagen type I hydrogels is their mechanical instability, which does not allow the production of larger gel-based transplants.
We report here a modified method that generates highly functional, mechanically stable skin substitutes, based on plastic compression (PC) of collagen type I hydrogels. 17 Due to the significantly higher density of collagen fibres, gel contraction and degradation are dramatically reduced. Autologous human fibroblasts can be evenly distributed in the three dimensions of the hydrogel and can be plastically compressed without affecting their viability and biological function.17,18 Notably, such skin substitutes can be prepared from small skin biopsies, and be successfully applied in one surgical intervention.19,20
Materials and Methods
Isolation and culture of human primary keratinocytes and fibroblasts
Human skin samples from foreskins were obtained after permission by the ethic commission of the Canton Zurich, and after written informed consent of the parents or patients. Skin biopsies were cut into small pieces of about 10 mm2 and digested for 15–18 h at 4°C in 12 U/mL dispase in Hank's balanced salt solution, containing 5 μg/mL gentamycin. The epidermis was mechanically separated from the dermis using forceps. Epidermal cells were isolated by incubation in 1% trypsin, 5 mM ethylenediaminetetraacetic acid for maximal 3 min at 37°C, and then resuspended in serum-free keratinocyte medium (SFM) containing 25 μg/mL bovine pituitary extract, 0.2 ng/mL epidermal growth factor, and 5 μg/mL gentamycin. Medium was changed every 2–3 days. Isolated cells were a mixture of mainly keratinocytes and melanocytes and were cultured in SFM.
The dermal tissue was digested in 2 mg/mL collagenase (Clostridiopeptidase A) for ∼60 min at 37°C. Isolated cells were seeded on 10 cm (diameter) cell culture dishes containing fibroblast growth medium (Dulbecco's modified Eagle's medium [DMEM] supplemented with 10% fetal calf serum [FCS], 4 mM
Culture of dermo-epidermal skin substitutes
Dermo-epidermal skin substitutes were prepared using a previously established transwell system (six-well cell culture inserts with membranes of 3.0 μm pore-size; BD Falcon, Basel, Switzerland). An acidic solution of bovine collagen type I (5 mg/mL; Symatese Biomatériaux, Chaponost, France; containing Neutral Red pH indicator) was neutralized on ice by dropwise addition of 0.5 M NaOH and immediately mixed with 5×104 human primary dermal fibroblasts (passage 1–2) suspended in DMEM. The total volume of the resulting gel was either 2 or 5 mL. The fibroblast containing gels were cultured in DMEM for 7 days before 7.5×105 keratinocytes were seeded on the surface of each gel. After initial submersed cultivation in Rheinwald and Green keratinocyte differentiation medium
21
(RGM: three parts DMEM and one part Ham's F12, 10% FCS, 4 mM
PC of round collagen hydrogels (2.3 cm in diameter)
T-shaped compression stamps (Fig. 1B, II) were constructed from polytetrafluoroethylene (PTFE) to fit into six-well filter inserts (BD Falcon). The length of the cylindrical part was chosen to obtain 0.5 or 1.0 mm thick compressed gels. For horizontal compression, a guiding bar was inserted into the compression stamp cylinder. Compression pressure and speed was adjusted using steel weight rings (Fig. 1B, I). Collagen gels containing 5×104 fibroblasts were prepared in cell culture six-well filter inserts as just described and incubated for 2 h at 37°C before compression. The filter inserts were transferred on sterilized filter paper (Whatman; VWR International, Dietikon, Switzerland), and the compression cylinder was inserted, loaded with 150 g weights, and the gels were compressed to their final thickness during 5 min. The stamp was removed with the gel remaining in the filter insert. The insert, containing the compressed gel, was transferred into the six-well dish after compression, and incubated in fresh fibroblast medium. Seven days after compression, keratinocytes were seeded on the gels according to the procedure just described.

Mechanical improvement of collagen gels by plastic compression.
Quantification of gel volume loss
To estimate the volume loss of collagen gels during cultivation in presence of fibroblasts, the wet weight of compressed and uncompressed gels was measured in a time course over 21 days after gel preparation. Collagen gels (5 mL) containing 5×104 human dermal fibroblasts were prepared and compressed to 0.5 mm as just described in six-well cell culture inserts. Uncompressed control gels (2 mL) containing 5×104 human dermal fibroblasts were prepared as previously described. 15 Before weighing, the medium was carefully aspirated from the gel and insert. The insert containing the gel was then weighed under sterile conditions, and then transferred back into fresh fibroblast growth medium. Gel wet weight was calculated by subtraction of empty insert weight. All weight measurements were performed on three gel replicates per condition, and gel weight loss at different time points was determined.
PC of square collagen hydrogels (7×7 cm)
To produce and compress square 7×7 cm collagen gels, we constructed a large compression chamber made from PTFE using a computerized numerical control (CNC) drilling machine (Fig. 4). A 0.5 mm thick sterilized filter paper (Whatman; VWR International) was placed into the perforated base plate and covered by a polyethylene terephthalate (PET) track-etched membrane with 5.0 μm pore size (Oxyphen AG, Lachen, Switzerland; Fig. 4A, B, I). Then, the casting frame (Fig. 4A, B, II) was clipped to the base plate to seal the chamber. 5×105 human primary dermal fibroblasts were diluted in 15 mL fibroblast growth medium, mixed well with 30 mL of bovine collagen Type I (5 mg/mL; Symatese Biomatériaux), and neutralized with 0.1 M NaOH on ice. The mixture was poured immediately into the chamber and incubated for 10 min at RT, then for 2 h at 37°C. For compression, the stamp (Fig. 4A, III) was inserted into the guiding bars, and weights were added. The final thickness of the compressed gels (0.6 mm) was determined by the stamp length. After 20 min of compression, the stamp was removed, and the gel was transferred into a 150 mm2 cell culture flask with reclosable lid (TPP, Trasadingen, Switzerland) containing fibroblast growth medium. The compressed gels were incubated floating in fibroblast growth medium at 37°C, 5% CO2. After initial cultivation for 7 days, 9×106 keratinocytes were seeded onto the gels, and the medium was switched to RGM. The medium was renewed every 1–2 days. Keratinocyte confluence, survival, and spreading was observed using fluorescein diacetate (FdA) live cell staining.
FdA live cell staining
FdA staining was performed as published and proved to be suitable for the determination of cell viability in tissue-engineered skin.22,23 In short, cell culture medium was replaced for 2 min with the equal volume of 5 μM FdA in PBS, freshly prepared from a stock solution of 5 mM FdA in acetone. FdA was removed by washing twice in PBS before fresh culture medium was applied. Overview pictures of fluorescein fluorescence were taken using an imaging system with epiblue exitation and fluorescein isothiocyanate (FITC) emission filter (G:BOX, Syngene, Cambridge, United Kingdom). Detailed views were acquired using a NIKON SMZ1500 fluorescent stereo microscope with FITC filters and a NIKON DXM1200F camera.
Transplantation of cultured dermo-epidermal skin substitutes
Immuno-incompetent female nu/nu rats (8–10 weeks old; Harlan, Horst, The Netherlands) were anesthetized with 15 mg/kg Ketamin (Ketalar®; Parke-Davis, Morris Plains, NJ), anaesthetised by inhalation of 5% Isofluran® (Baxter, Volketswil, Switzerland), and maintained by inhalation of 2.5% Isofluran via mask. For eye protection, Viscotears® cream (Novartis, Bern, Switzerland) was applied. Round gel samples were cut with a punch, 26 mm in diameter, and transplanted onto full-thickness skin defects created surgically on the back of the animals and encased by polypropylene rings, 26 mm in diameter (modified Fusenig chamber 24 ). The transplants were covered with a silicon foil. After operation, 0.5 mg/kg buprenorphine (Temgesic®; Essex, Luzern, Switzerland) was injected subcutaneously. The animals were sacrificed by CO2 application 21 days after transplantation. Grafts were excised in toto, embedded in optimal cutting temperature compound (Tissue-Tek®; Sakura Finetek, Japan), and 12 μm thick cryosections were prepared. For histological analysis, sections were stained with haematoxylin and eosin (H&E; Sigma, St. Louis, MO) and thereafter mounted within Eukitt® (Fluka, Buchs, Switzerland).
Antibodies
The following primary antibodies were used for immunostaining: K1 (clone LHK1, 1:200; Novus Biologicals, Littleton, CO); K15 (clone LHK15, 1:100; R&D Systems, Abingdon, United Kingdom); K19 (clone RCK108, 1:100; Santa Cruz, Labforce AG, Nunningen, Switzerland); Ki67 (clone B56, 1:200; ABD Serotec, Dusseldorf, Germany); Laminin-332 (clone P3H9-2, 1:100; Chemicon, Millipore AG, Zug, Switzerland); human CD90/Thy-1 (clone AS02, 1:100; Calbiochem, Dietikon, Switzerland, VWR International); CD31 (clone TDL-3A12, 1:50; BD Pharmingen, Allschwil, Switzerland); melanosome (clone HMB45, 1:50; Dako, Baar, Switzerland); integrin α6-PE (clone GoH3, 1:50; BD Pharmingen). Actin filaments were stained using Phalloidin-TRITC 0.1 μM (Sigma).
For immunofluorescence, the primary antibodies were preincubated with Alexa 555 or Alexa 488 conjugated polyclonal goat F(ab’)2 fragments, according to the instructions of the manufacturer (Zenon Mouse IgG Labeling Kit, Molecular Probes, Invitrogen).
Immunofluorescence staining
Cryosections (12 μm) were fixed and permeabilized in acetone for 5 min at −20°C, air dried and washed 3× in PBS. After blocking for 30 min in 2% BSA in PBS (Sigma), the antibody mixtures were added in blocking buffer for 1 h at room temperature. Slides were washed for 5 min in PBS, nuclei stained for 5 min with 1 μg/mL Hoechst 33342 (Sigma) in PBS, and then washed twice for 5 min in PBS. Finally, the samples were mounted with Dako fluorescence mounting solution (Dako).
Fluorescence microscopy
Fluorescence microscopy pictures were acquired using a DXM1200F digital camera connected to a Nikon Eclipse TE2000-U inverted microscope equipped with Hoechst, FITC, and TRITC filter sets or Nikon SMZ1500 stereo microscope with FITC filters (Nikon AG, Egg, Switerland; Software: Nikon ACT-1 vers. 2.70).
Images were processed with Photoshop 7.0 (Adobe Systems Inc., Munich, Germany).
Electron microscopy
All electron microscopy procedures were performed at the Center for Microscopy and Image Analysis of the University Zurich, Switzerland, according to the center's general protocols.
Tissue samples for transmission electron microscopy (TEM) were prefixed in 0.1 M cacodylate buffer (Merck, Hohenbrunn, Germany), pH 7.3 containing 2.5% glutaraldehyde for 2 h. After washing in cacodylate buffer, the samples were postfixed in 1% OsO4 and 1.5% K4Fe(CN)6 for 1 h, then dehydrated in a graded series of ethanol, and embedded in EPON 812 (Catalys AG, Wallisellen, Switzerland). Approximately 50–70 nm ultra thin sections were transferred onto copper grids and contrasted with 4% uranyl acetate and 3% lead citrate. The photographs were acquired using a CM 100 transmission electron microscope (Philips, Eindhoven, The Netherlands). Where not indicated differently, reagents were from Sigma.
For scanning electron microscopy (SEM), samples were fixed in 3% glutaraldehyde in PBS for 40 min, and then washed thrice in PBS, followed by postfixation in 2% Osmiumtetroxid for about 20 min. After three washes in distilled water, the samples were dehydrated in a graded series of ethanol, then washed thrice in absolute ethanol, and dried in a Critical-Point-Dryer. The dried samples were glued on SEM stubs and coated with Platinium. Pictures were acquired using a Zeiss Supra 50 VP scanning electron microscope. All reagents were from Sigma.
Results
Defined PC improves mechanical properties of collagen gels
The drawback of uncompressed collagen type I hydrogels is their mechanical instability (Fig. 1A). We have now developed two different hydrogel compression devices, which allow PC of collagen hydrogels, and, hence, the production of mechanically resistant, dermo-epidermal skin substitutes to be used both experimentally and clinically.
One of the compression devices is compatible with commercially available six-well cell culture membrane inserts (Fig. 1B). Its compression stamp was designed to stop compression at a defined thickness of 0.5 mm to create a real three-dimensional (3D) matrix rather than a collagen foil. Collagen gels (11 mm thick) were prepared in six-well cell culture membrane inserts and compressed to 0.5 mm. Liquid drain flux through the membrane was sufficient to allow compression within 3–5 min when loaded with 150 g weights. The collagen type I concentration in compressed hydrogels was ∼65 mg/mL. The resulting gels were mechanically stable with significantly improved handling properties in comparison to uncompressed gels (Fig. 1A, C).
Electron microscopy of plastically compressed collagen hydrogels
TEM revealed that typical striated collagen fibres were detectable in both compressed and uncompressed gels (Fig. 2A, B). However, fiber concentration was significantly higher in compressed gels (Fig. 2B). Collagen fibrrs were more aligned parallel in compressed gels (Fig. 2B). These observations are in line with previous reports on collagen fiber organization in compressed gels. 25
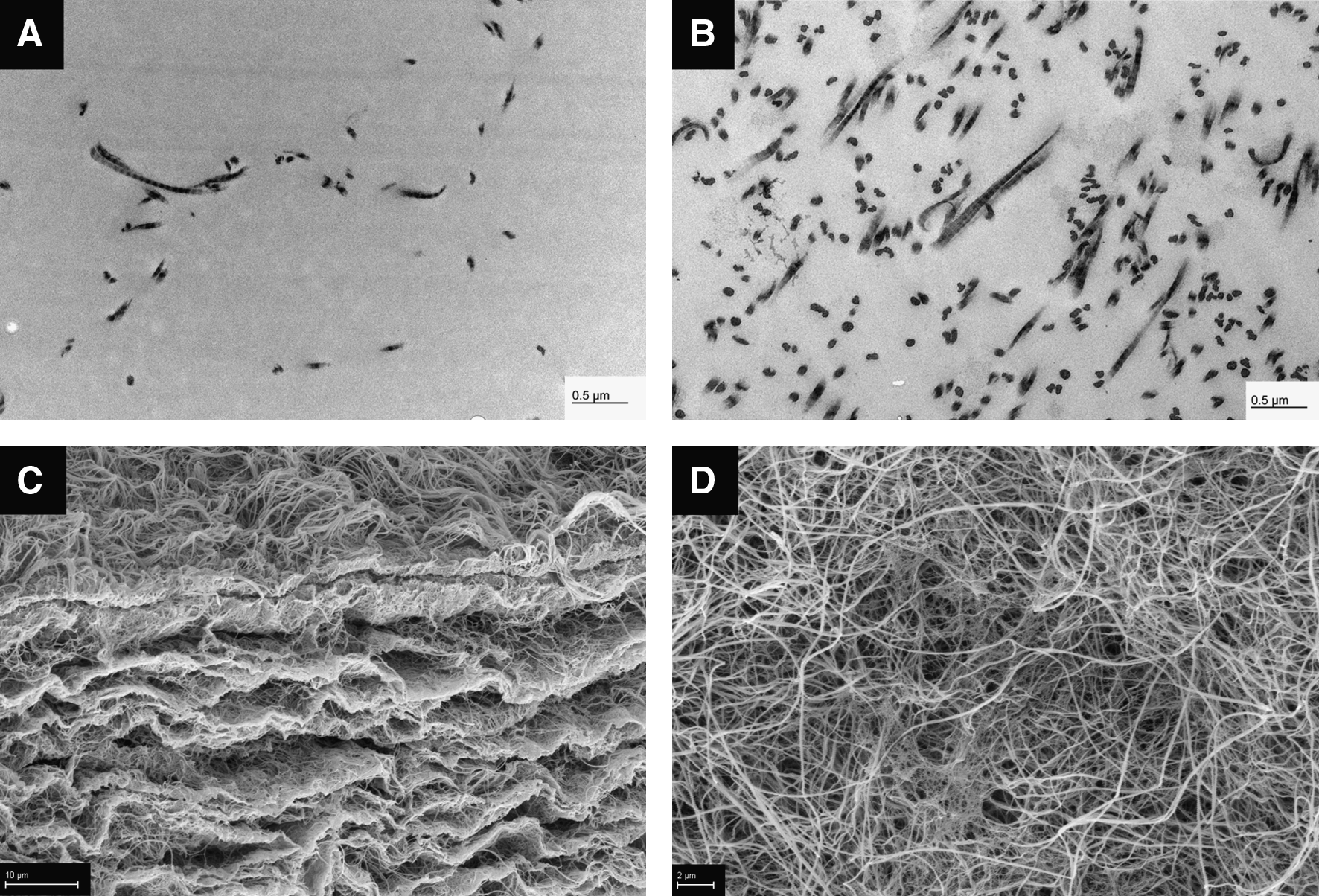
Electron microscopy of compressed collagen gels. TEM showing striated collagen fibres in
SEM revealed that compressed collagen gels contained several layers of parallel dense collagen layers that were interconnected by a loose collagen network (Fig. 2C). The top view of the gels showed a dense collagen network with randomly orientated collagen fibers (Fig. 2D).
Defined compression improves biological stability of collagen gels
A major drawback of uncompressed collagen gels is their tendency to lose volume when seeded with dermal fibroblasts. 26 Volume loss can be caused by gel self-compression, contraction, or collagen degradation, induced by fibroblasts during the process of cell-mediated ECM modulation.17,26–28
To assess a time course of volume loss, we measured the gel's wet weight, as weight is proportional to volume (assuming constant mass density). Both compressed and uncompressed gels, seeded with 5×104 human primary dermal fibroblasts, were cultured under submersed conditions, and weighted during a time course of 21 days. Uncompressed gels (2 mL) lost more than 60% of their original weight during the 21 day cultivation period, whereas 5 mL gels compressed to 0.5 mm kept their original weight almost constant during the same time (Fig. 3). Of note, pronounced wet mass loss was typically accompanied by irregular surface area reduction (contraction, data not shown); therefore, our results are in line with the already reported reduction of gel surface area loss after PC. 29

Time course of gel wet weight loss: during a 21 day cultivation period, 5 mL collagen type I hydrogels seeded with fibroblasts and compressed to 0.5 mm thickness (open boxes) lost <15% of their original wet weight. Uncompressed gels (black rhombi) lost >60% under the same conditions. Error bars indicating standard deviation (n=3).
PC of square hydrogels (7×7 cm)
To generate dermo-epidermal skin substitutes in a clinically relevant size, we developed a gel casting and compression device and established culture conditions that allowed the growth of larger, 7×7 cm dermo-epidermal skin substitutes.
The casting/compression device was developed by utilizing computer-aided design software (CAD), then cut from PTFE using a CNC drilling machine. As depicted in Figure 4A and B, the new device consisted of four major parts: (I) a perforated base plate onto which a filter paper and a porous PET track etched membrane were placed; (II) a casting frame that was clipped and sealed to the base plate; (III) a compression stamp that fitted into the casting frame and was vertically guided by four steel bars; (IV) adjustable weights to be placed onto the stamp to define the compression force. All parts can be easily disassembled for cleaning and sterilising. Using distinct stamp lengths, it was possible to exactly determine the final thickness of the compressed gel.

Compression of 7×7 cm collagen gels.
Square hydrogel (7×7 cm) was cast into the device and compressed to 0.6 mm, 2 h after gelation. Importantly, the gel could be poured, gelled, and compressed without the need to dislocate the mechanically weak uncompressed gel. After compression, the gels obtained strong mechanical stability, so that lifting with forceps and transfer into culture dishes was easily possible (Fig. 4C).
Generation of dermo-epidermal skin substitutes using defined collagen gel compression
Fibroblast-containing, compressed hydrogels did not settle down to the cell culture dish. Therefore, floating cultivation without any mechanical support was possible.
FdA vital staining confirmed fibroblast survival in compressed gels (data not shown). Beta-actin staining using Phycoerythrin conjugated Phalloidin, followed by confocal microscopy, showed the formation of 3D networks of fibroblasts, already 5 days after gel compression (Fig. 5A). This fibroblast network was stable for at least 3 weeks (data not shown).
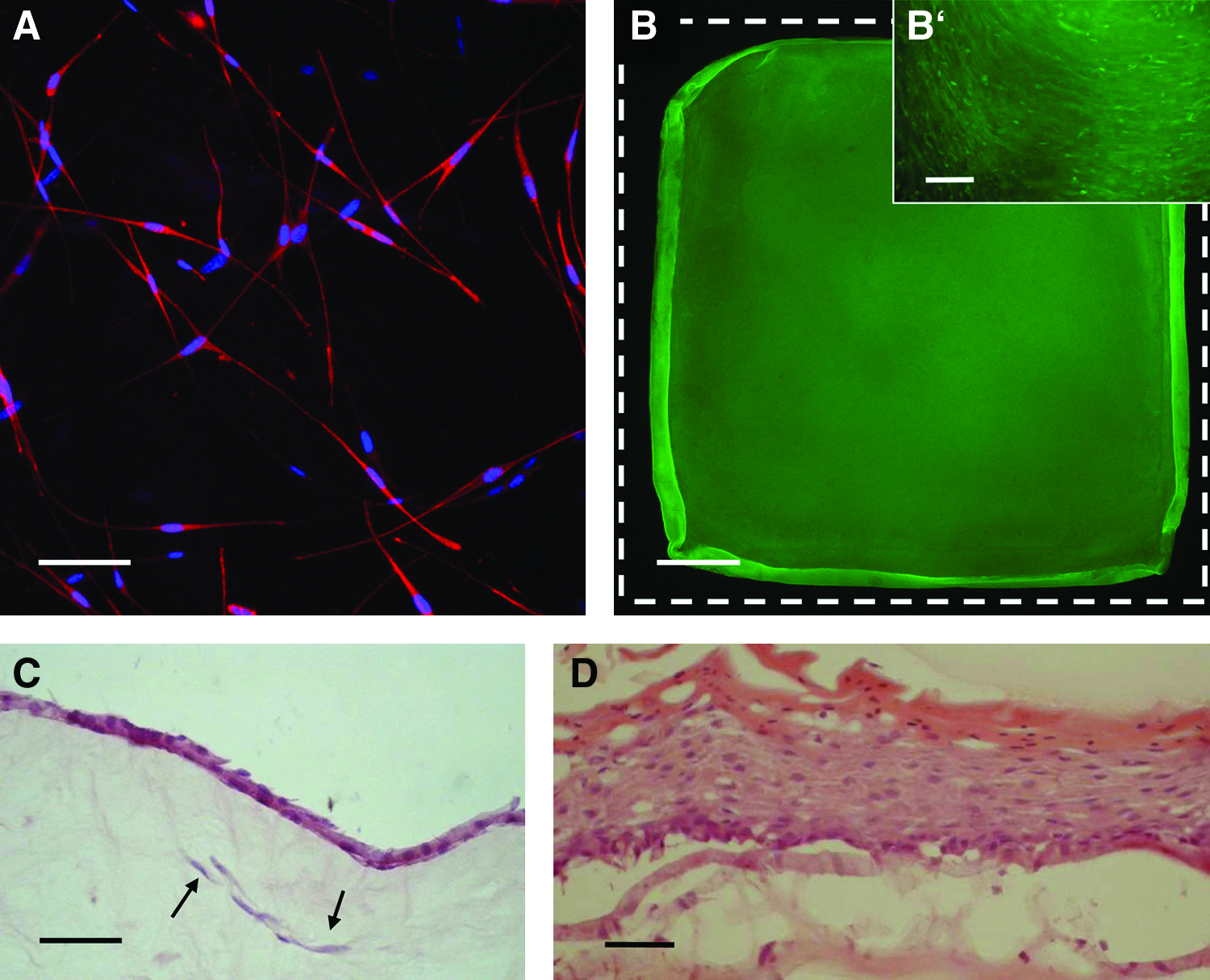
In vitro cultivation of square compressed gels.
Seven days after compression, keratinocytes were seeded at a high density onto one surface of these gels. Thereafter, the dermo-epidermal skin substitutes were cultured for additional 7 days, freely floating in medium. At this stage, FdA staining confirmed that the entire upper surface of the gel was covered by a confluent keratinocyte layer (Fig. 5B, B’). Cultured compressed gel-based, dermo-epidermal substitutes did not deform or shrink during this 14 day incubation period.
H&E staining revealed a continuous keratinocyte layer with limited stratification (two to three layers—a stratum corneum could not yet be observed) at this time, as expected (Fig. 5C). Importantly, healthy fibroblasts were still detectable in the gels. Since square skin substitutes, 7×7cm in size, can neither be cultured at the air-liquid interphase, nor transplanted onto immune incompetent rats, we cut round sample areas, 2.6 cm in diameter, from the 7×7 cm skin substitutes and processed them for subsequent in vitro and in vivo experiments. For in vitro stratification, these samples were cultured for an additional 10 days in six-well cell culture inserts at the air-liquid interphase. H&E staining revealed a multilayered cornified epidermis (Fig. 5D).
In vivo studies
For in vivo studies, samples were transplanted on full thickness wounds onto immuno-incompetent rats. Within 3 weeks after transplantation, the skin substitutes developed a homogenous skin-like appearance on the entire transplant area (Fig. 6A). H&E staining revealed a near normal skin morphology with a multilayered (stratified) cornified epidermis on top of a fibroblast containing dermal component (Fig. 6B).
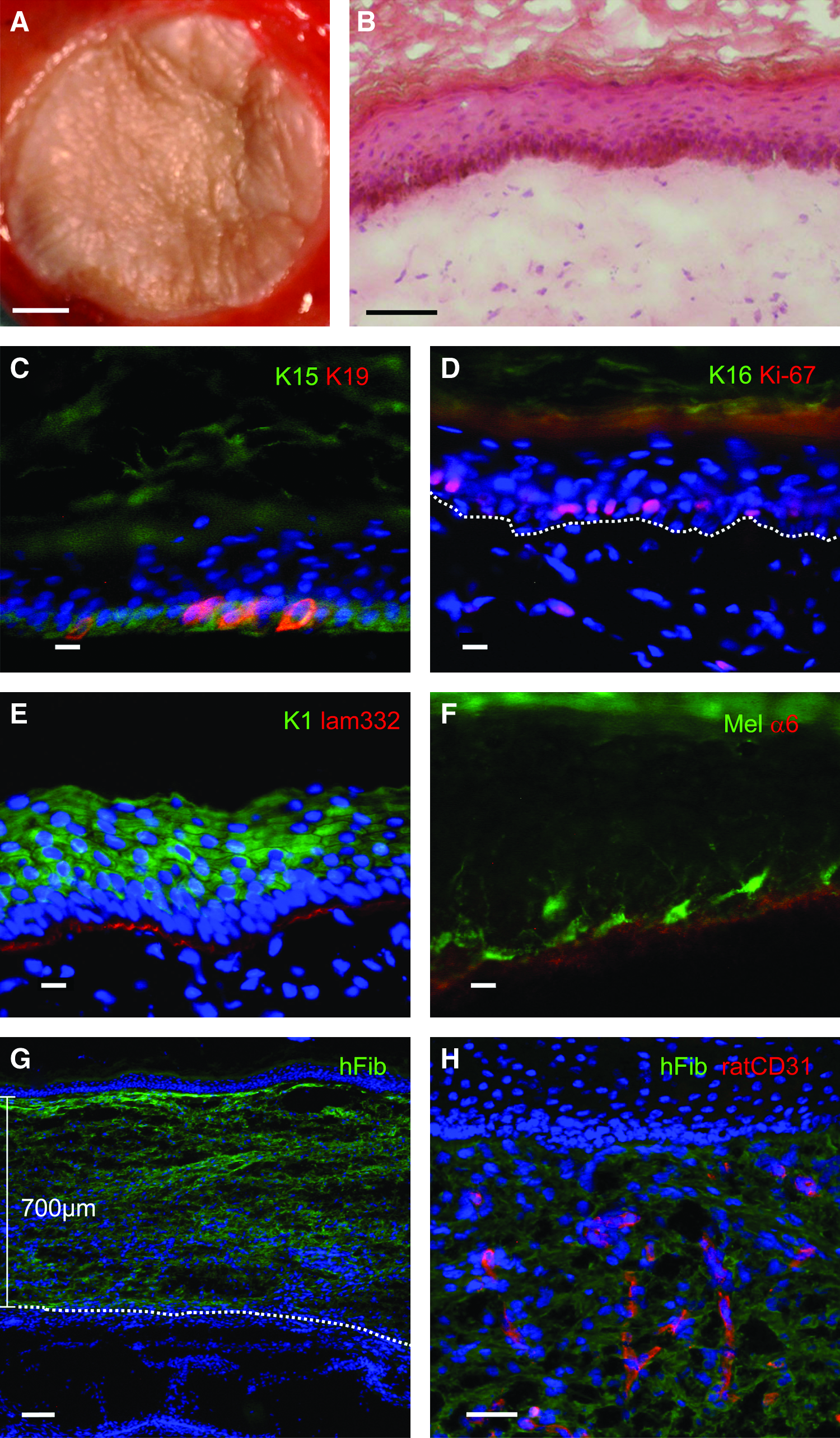
Transplantation of 5.2 cm2 biopsies from 7×7 cm engineered skin substitutes.
The quality of the transplanted skin substitutes was evaluated by immunofluorescence staining employing a set of established markers for skin homeostasis and biological function.15,30–33 Keratin 15 (K15) is a marker for basal keratinocytes anchored to a functional basement membrane in young normal interfollicular skin and indicates skin homeostasis. The majority of basal keratinocytes expressed K15, while no suprabasal K15 expression was detectable 3 weeks after transplantation (Fig. 6C).
An additional strong indicator for epidermal homeostasis is the expression of keratin 19 (K19) occurring exclusively in basal keratinocytes. 15 Three weeks after transplantation, K19 expression was restricted to a subset of basal, K15-positive keratinocytes, thus resembling the situation of a healthy young skin (Fig. 6C).
Keratin 16, exclusively expressed in a wound healing situation34–36 was no longer detected in the central part of the transplant (Fig. 6D). Additionally, cell proliferation was restricted to basal keratinocytes, as shown by the cell proliferation marker Ki-67 37 (Fig. 6D).
A reliable indicator for keratinocyte differentiation is Keratin 1 expression throughout all suprabasal epidermal layers (Fig. 6E). Using laminin-332 antibodies, the onset of basal lamina deposition was detected (Fig. 6E).
Melanocytes, which were co-isolated and cultured with the keratinocytes, survived on compressed gels and were evenly distributed in the basal epidermal layer, with extensions reaching into higher strata (Fig. 6F). As also shown in this figure, basal keratinocytes expressed the α6 chain (Fig. 6F) of integrin α6ß4, the receptor for laminin-332.
Immunostaining using a human specific fibroblast antibody (anti-human CD90) showed the human origin of dermal fibroblasts in the transplant, whereas the underlying rat tissue was not stained (Fig. 6G). The thickness of the human-derived neodermis was 0.7 mm in nonfixed cryosections and, therefore, close to the original thickness of the transplanted gel (0.6 mm). Within this human dermis, rat vascular structures could be detected using CD31 antibodies (Fig. 6H), indicating that the compressed hydrogels obviously allowed for efficient vacularization. In summary, it can be said that all markers investigated showed expression patterns closely resembling young and functional skin.
Scanning electron microscopy
The immuno-fluorescence data just mentioned are supported by SEM analysis (Fig. 7). The stratum corneum covering the entire epidermal surface appeared very similar to the stratum corneum of normal human skin (sc in Fig. 7A). Freeze fracture SEM (Fig. 7B) revealed that the epidermis (ed) consisted of several layers of tightly packed keratinocytes, with prominent spherical nuclei (white arrowheads). In the dermis (de), a dense network of collagen fibres was clearly visible. Within the dermis, several blood vessels containing erythrocytes (white arrows) were detectable. Compared with hydrogels without seeded fibroblasts (Fig. 2C), cultured and transplanted engineered skin substitutes showed a largely altered ECM, exhibiting a denser and finer fibrillar network, thus indicating an active ECM modification by the cells.

SEM of dermo-epidermal skin substitute 3 weeks after transplantation.
Discussion
A crucial precondition for the production of complex (dermo-epidermal) skin substitutes is the extracellular matrix providing a substrate for all cell types involved. Collagen type I hydrogels are ideal in supporting the 3D distribution of fibroblasts within the gel, and the development of a continuous and evenly stratified epidermis on the gel.11,15 However, collagen gels are mechanically weak and cannot be safely handled in large sizes. We modified the originally described method of PC 17 to generate mechanically resistant collagen hydrogels in clinically relevant sizes, to be used in combination with primary dermal and epidermal skin cells. Importantly, the resulting skin grafts can be securely and conveniently handled by the surgeon. Our modified hydrogel compression procedure is simple and reliable. It may be automated for serial production of full-thickness skin grafts or for the generation of skin grafts used for high throughput testing.
There are two important new features of the described compression procedure: (1) Casting and compression of the hydrogel can be done within the same vessel, and thus dislocation of the gel is not required. The small and round type of skin substitutes can even be cultured in the same vessel. (2) The final thickness of the gels generated by the method described here can be exactly defined. We used hydrogels compressed to the thicknesses of 500, 600, and 1000 μm, respectively. This is in contrast to the gel compression described by Brown et al., which varied in thickness at around 60 μm. 17
Of course, the effect of modified PC on fibroblasts, keratinocytes, and the potency to generate a dermo-epidermal skin graft in vitro and in vivo is of pivotal importance. Our data show that human primary dermal fibroblasts seeded into collagen hydrogels survive PC, remain biologically active, exhibit a spindle-like morphology, and form 3D networks. This is in accordance to previous publications, reporting on the effects of PC on human dermal fibroblasts, 17 murine dermal fibroblasts, 29 human limbal fibroblasts 18 and human smooth muscle cells.38,39
We found that the dermal compartment of the transplants was always well developed (700 μm in thickness) and entirely intact, 3 weeks after transplantation. Keratinocyte proliferation and differentiation (stratification) is highly dependent on such a functional dermis, because an epidermis without the support of functional dermal fibroblasts would not survive.26,40,41 Apparently, these fibroblasts are permanently producing a set of biologically active factors at physiological concentrations. The development of a naturally stratified and fully functional epidermis on compressed collagen gels is, therefore, a reliable indicator for the fruitful interaction between dermal fibroblast and epidermal keratinocytes. The basis for this profitable biological interaction is provided by the thickness, density, and consistency of the corresponding collagen hydrogel.
Conclusions
We show here that compressed collagen hydrogels are excellent matrices to enable successful in vitro and in vivo skin reconstitution. Importantly, no negative biological effects of compression are observed. The mechanical stability of compressed collagen gels is by far superior to those of uncompressed gels, while their admirable biological properties remain unaltered by the compression process. In essence, it can be said that the method of PC of collagen type I hydrogels, and their organ-typic cellular population, make now the clinical application of this type of skin substitutes possible.
Footnotes
Acknowledgments
This work was financially supported by EU-FP6 project EuroSTEC (soft tissue engineering for congenital birth defects in children: contract: LSHB-CT-2006-037409) and by the University of Zurich. The authors are particularly grateful to the Fondation Gaydoul and the sponsors of “DonaTissue” (Thèrèse Meier, Robert Zingg, the Vontobel Foundation, and the Werner Spross Foundation).
The authors are very thankful for the great support in constructing and producing the compression devices by Daniel Bollier, Institute of Plant Biology, University of Zürich.
Disclosure Statement
No competing financial interests exist.