
Abstract
Development and implementation of therapeutic protocols based on stem cells or tissue-engineered products relies on methods that enable the production of substantial numbers of cells while complying with stringent quality and safety demands. In the current study, we aimed to assess the benefits of maintaining cultures of adipose-derived stem cells (ASCs) in a defined culture system devoid of xenogeneic components (xeno-free) and hypoxia over a 49-day growth period. Our data provide evidence that conditions involving StemPro mesenchymal stem cells serum-free medium (SFM) Xeno-Free and hypoxia (5% oxygen concentration) in the culture atmosphere provide a superior proliferation rate compared to a standard growth environment comprised of alpha-modified Eagle medium (A-MEM) supplemented with fetal calf serum (FCS) and ambient air (20% oxygen concentration) or that of A-MEM supplemented with FCS and hypoxia. Furthermore, a flow cytometric analysis and in vitro differentiation assays confirmed the immunophenotype stability and maintained multipotency of ASCs when expanded under xeno-free conditions and hypoxia. In conclusion, our data demonstrate that growth conditions utilizing a xeno-free and hypoxic environment not only provide an improved environment for the expansion of ASCs, but also set the stage as a culture system with the potential broad spectrum utility for regenerative medicine and tissue engineering applications.
Introduction
The use of ASCs for regenerative applications is subject to particular local health authority regulations, and it is therefore desirable that the highest possible standards are achieved. From that perspective, it reasonable that the paradigm is shifting from procedures that involve growth environments comprised of nonhuman animal biological products to systems that are defined and devoid of xenogenic components (xeno-free). Recently, defined serum-free and serum-free/xeno-free commercial products have been introduced that appear to perform favorably in terms of mesenchymal stem cell proliferation rates and maintained multipotency compared to traditionally used fetal calf serum (FCS)-supplemented media.11,12 In addition to the biochemical environment, it has recently been shown that hypoxia plays an important regulatory role and has the potential to modulate both the proliferative capacity and self-renewal in several types of stem cells, including embryonic stem cells (ESCs),13,14 limbal stem cells,15–17 as well as neural18,19 and myoblastic20,21 precursors. Mesenchymal stem cells (MSCs) are also subject to the regulatory effects of a hypoxic atmosphere and have been shown to grow at a faster rate along with a well-preserved differentiation potential under such conditions.22–24
Cell-based therapeutic applications often require very large batches of cells, with doses ranging from 107 to 108 cells (
Materials and Methods
Cell culture
Cells were isolated as described previously.22,24,25 Three lines of ASCs, designated ASC12, 21, and 23, which have been isolated and characterized previously, 23 were continually grown in standard growth conditions involving an alpha-modified Eagle medium (A-MEM; Invitrogen, Carlsbad, CA) supplemented with 10% FCS (Europa Bioproducts Ltd., Cambridge, United Kingdom), 100 IU/mL penicillin, 100 μg/mL streptomycin, and 50 μg/mL gentamicin or in defined and xenogeneic-free conditions involving the StemPro® MSC SFM XenoFree+CELLstart™ culture system (StemPro) (all from Invitrogen). The essential feature of StemPro medium is that it is supplemented with a platelet-derived growth factor, transforming growth factor (TGF)-β, and fibroblast growth factor. 26 The medium is based on a proprietary formulation, but further information can be obtained directly from the manufacturer. The cultures were initiated in duplicate at 2000 cells per cm2 in T25 tissue culture flasks (Costar, Acton, MA) and further serially passaged at 1-week intervals. The medium was changed between passages once for A-MEM and three times for StemPro. For hypoxic expansion, the cell cultures were placed in an Xvivo System contained hypoxic workbench/incubator (BioSpherix, Redfield, NY). The gas conditions involved 5% concentrations of both the oxygen and carbon dioxide that was buffered with nitrogen. Cell propagation was carried out while maintaining the cells strictly within the hypoxic confinement of the workstation to avoid reoxygenation. The doubling time (Td) was calculated according to the equation Td=(T2−T1)log2/log(Q2/Q1), where T is time and Q is quantity of cells, and the total number of population doublings (Nd) was determined using the formula Nd=log(Q2/Q1)/log2.
Flow cytometric analysis
ASCs were released from monolayer cultures using a successive treatment with collagenase H1 (Wako, Neuss, Germany) and trypsin (Invitrogen). After inactivation of trypsin with A-MEM+ 10% FCS, the suspension was filtered through a cell strainer (70 μm; BD Biosciences, Brøndby, Denmark), and ∼2×105 cells were aliquoted into eppendorf tubes. Following two washing steps with 2% FCS and 0.1% sodium azide in phosphate buffered saline (PBS) (FBS) at 300 g for 3 min, the cell pellets were resuspended in 15 μL of F-PBS. The cells were then reacted with antibodies (0.5 μg) that were prelabeled using the Zenon technology (Invitrogen) at room temperature for 30 min. The free fluorophore complexes were removed by two more washing steps, and the suspensions were fixed with 1% buffered formaldehyde. Zenon 488, PE, and 647 primary antibody conjugation systems allowed the simultaneous detection of three markers. Primary antibodies included CD24, CD29, CD73, CD117, CD133, CD146, CD200, CD271, and HLA DR (all from Abcam, Cambridge, United Kingdom); CD34, CD44, CD45, CD105, and HLA ABC (all from DAKO, Glostrup, Denmark); CD90 and ABCG2 (both from Santa Cruz Biotechnology, Heidelberg, Germany); and Stro-1 (Millipore Chemicon, Copenhagen, Denmark). Matching type and isotype antibodies were used in control samples. The proportion of live cells was determined by using the Live/Dead Reduced Biohazard Cell Viability Kit #1 (Invitrogen) according to manufacturer's instructions.
After having performed the calibration and compensation procedures, the cell populations were analyzed using a FACSCanto flow cytometer (BD Biosciences) by acquiring at least 1×104 events, and the data were processed using BD FACSDiVa (BD Biosciences) and FlowJo (TreeStar, Ashland, OR) software packages. Gating based on bivariate forward versus side scatter plots was used to eliminate cellular debris, and the threshold for positive events was determined as the upper 5 percentile of intensity distribution in the control samples.
Differentiation assays
Adipo-, osteo-, and chondrogenic differentiation assays were carried out according to previously published protocols22,27,28 with minor modifications. Briefly, for adipo- and osteogenic induction, cultures were initiated at a density of 300 cells/cm2 in 24-well plates and maintained in a standard growth medium for ∼14 days until they reached confluency. For chondrogenic induction, a high-density pellet culture format as previously described by Penick et al. 29 was used. The cultures were comprised of 2×105 cells pelleted at 500 g for 5 min in 96-well V-bottom plates (Corning, Schiphol-Rijk, The Netherlands) in a standard growth medium. All cultures were kept in a humidified atmosphere containing 5% CO2 buffered with ambient air at 37°C.
To initiate adipogenic differentiation, the growth medium was replaced with an induction cocktail consisting of Dulbecco's modified Eagle's medium (DMEM)/F12 supplemented with 10% FCS, 0.1 μM dexamethasone, 0.45 mM isobutylmethylxanthine, 170 nM insulin, 0.2 mM indomethacin, antibiotics (all from Sigma-Aldrich, Brøndby, Denmark), and 1 μM rosiglitazone (Cayman Chemicals, Tallin, Estonia). After 2 weeks, the cultures were fixed with 4% formaldehyde, and the presence of lipid droplets was assessed by staining with 3% oil red O. To initiate osteogenic differentiation, the growth medium was replaced with an induction medium consisting of DMEM/F12 supplemented with 10% FCS, 0.1 μM dexamethasone, 50 μM L-ascorbic acid 2-phosphate, 0.5 μM calcitriol, 10 mM glycerol 2-phosphate, and antibiotics (all Sigma-Aldrich). After 4 weeks, the cultures were fixed with 70% ethanol, and the degree of induction was evaluated by staining for calcium deposits with 2% alizarin red. For chondrogenic induction, a high-glucose (4.5g/L) DMEM supplemented with 10 ng/mL TGF-β3 (RnD Systems, Oxon, United Kingdom), 10−7 M dexamethasone, 50 μg/mL L-ascorbic acid 2-phosphate, 40 μg/mL L-proline, antibiotics and an insulin, selenium, and transferrin supplement (ITS; BD Bioscience) was used. After 3 weeks, the pellets were fixed with 4% formaldehyde and further processed using standard procedures for paraffin embedding and sectioning. Accumulation of glycosaminoglycans was detected by staining with Alcian blue 8GX, and the sections were counterstained with Mayer's hematoxylin (Bie & Berntsen, Herlev, Denmark). The microscopic evaluation and recording were done with the Zeiss Axiovert 200M inverted microscope using the Zeiss AxioVision software package (Brock & Michelsen, Birkerød, Denmark).
For all differentiation assays, control cultures were initiated where cells were cultured in a manner similar to the differentiation studies, however, in normal growth medium.
Real-time reverse transcriptase–polymerase chain reaction
Total RNA was isolated using the Aurum Total RNA mini kit (Bio-Rad, Copenhagen, Denmark) according to manufacturer's instructions. Yield and purity were determined electrophoretically (Agilent 2100 Bioanalyzer; Agilent Technologies, Nærum, Denmark), and cDNA was synthesized from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad).
Quantitative polymerase chain reaction (PCR) was performed on a My-Cycler real-time PCR system (Bio-Rad). The reactions were carried out in duplicates in a final volume of 25 μL, comprised of SYBR Green PCR supermix (Bio-Rad), 0.25 μL cDNA, and 5 pmol of the primers. Primer sequences were designed using the PrimerSelect application within the Lasergene software package (DNA-STAR, Madison, WI) or adopted from previous work.30,31 All primers were manufactured by DNA Technology (Aarhus, Denmark) and included the following:
Lineage-specific genes: PPARG2 forward 5′-TCA GGT TTG GGC GGA TGC-3′ and reverse 5′-TCA GCG GGA AGG ACT TTA TGT ATG-3′, RUNX2 forward 5′-GGC AGC ACG CTA TTA AAT CC-3′ and reverse 5′-GTC GCC AAA CAG ATT CAT CC-3′, and SOX9 forward 5′- TTC GGT TAT TTT TAG GAT CAT CTC G-3′ and reverse 5′- CAC ACA GCT CAC TCG ACC TGG-3′.
Reference genes: tyrosine 3/tryptophan 5-monooxygenase activation protein (YWHAZ) forward 5′-ACT TTT GGT ACA TTG TGG CTT CAA-3′, reverse 5′-CCG CCA GGA CAA ACC AGT AT-3′ and cyclophilin A (PPIA) forward 5′-TCC TGG CAT CTT GTC CAT G-3′ and reverse 5′-CCA TCC AAC CAC TCA GTC TG-3′.
The thermal cycling protocol consisted of initial denaturation for 3 min at 95°C and was followed by 45 amplification cycles of 15 s at 95°C and 30 s at the annealing and extension temperature of 68°C for PPARG2 and 60°C for all other genes. To confirm product specificity, a melting curve analysis was performed following each amplification cycle. The relative expression level for each gene was calculated on the basis of a fourfold serially diluted standard curve derived from a pool of all the cDNA samples. The values of the lineage-specific genes were normalized to the geometric mean of the values of YWHAZ and PPIA. In each assay, a blank without cDNA was included as a control.
Statistical analysis
Data are presented as a mean±standard error of mean (SEM). The pairwise comparison of sample means was carried out using nonparametric statistics, including Mann-Whitney U and Kolmogorov-Smirnov tests. The statistical significance was assigned to p<0.05. The procedures were carried out with the aid of SPSS 17 software package (SPSS, Chicago, IL).
Results
Effect of growth media and oxygen concentration on proliferation rates and morphology of ASCs
After primary isolation from fat tissue (P0) in an A-MEM growth medium and under standard atmospheric conditions (5% CO2, 20% O2), the ASC12, 21, and 23 lines were expanded for two additional passages (P2). At this point, all lines were seeded in test conditions, including A-MEM, under hypoxic (5% CO2, 5% O2) or control (5% CO2, 20% O2) atmospheric conditions or in StemPro in hypoxic conditions for an additional 7 passages (P9 in total, 49 days). Analysis of cumulative cell expansion revealed that ASCs in StemPro+5% O2 displayed enhanced cell growth compared to cells grown in A-MEM under control atmospheric conditions (Fig. 1A). At the end of the 49-day expansion period, the cumulative cell numbers for StemPro+5% O2 surpassed nearly 250-fold those under control conditions (1503.3E+8 vs. 6.0E+8, respectively). Also, the number of cells grown in A-MEM+5% O2 was significantly higher at the end (29-fold) compared to control atmospheric conditions. By normalizing the growth in hypoxia, together with the results from StemPro+20% O2 (data not shown), to the values from control growth conditions, a measure of relative contribution for each condition and their combination can be obtained. Thus, the StemPro (StemPro+20% O2) and hypoxic (A-MEM+5% O2) conditions contributed to a 36- and 9-fold increase, respectively, whereas the combination of StemPro and hypoxia resulted in a 167-fold increase of the cell numbers. Consequently, these data indicate that the StemPro and 5% O2 have a capacity to boost the cell proliferation in a synergistic fashion. An analysis of total Nd and Td (Table 1) similarly revealed that ASCs grown in StemPro+5% O2 yielded significantly higher (1.3- and 1.6-fold) total Nd values compared to the cultures in A-MEM+5% O2 or control atmospheric conditions, respectively.

Effect of growth media and hypoxic culture atmosphere on expansion rate of human ASCs.
Three lines, ASC12, 21, and 23, were analyzed.
Statistically significant difference (p<0.05) from A-MEM at both oxygen concentrations.
Statistically significant difference (p<0.05) from A-MEM 20% O2.
Td, doubling time; Nd, population doublings; ASC, adipose-derived stem cell; A-MEM, alpha-modified Eagle medium.
The difference in total numbers is evident in StemPro+5% O2 cultures as early as the first passage, reflecting the high proliferative rate (Fig. 1A). It is noteworthy that the hypoxic cultures grew in an apparently bi-modal pattern, with significantly accelerated growth observed during the first 28 days for A-MEM+5% O2 compared to control conditions and with StemPro+5% O2 revealing superior performance compared to A-MEM (Table 1). In the latter part of the culture period, the growth rates under hypoxic conditions (A-MEM and StemPro) slowed, although growth rates were still equal or greater than control conditions, where the Td appeared to remain consistent for the duration of the experiment. Despite a slowdown during the last 3 weeks, the overall proliferation rate in StemPro+5% O2 was significantly higher than in the control condition. Only in StemPro+5% O2, the cells grew with a significantly different rate throughout the entire growth period.
Regarding morphology, cells grown under all conditions maintained a characteristic ASC appearance (Fig. 1B). However, under more detailed scrutiny, when grown in StemPro+5% O2, the cells appeared more compact and spindle-shaped with highly refractive edges and with a less extensive cytoplasmic area. There were no differences in morphological features across different oxygen concentrations in the A-MEM cultures. Moreover, when the cells grown in StemPro in control atmospheric conditions were examined, they exhibited a pattern similar to their counterparts grown in hypoxic conditions (data not shown).
Stability of ASC immunophenotype during long-term expansion in different growth media and oxygen concentrations
To assess temporal phenotypic changes of ASCs grown under different media and atmospheric conditions, cells freshly isolated (P0) and those expanded for 49 days (P9) were examined. The comprehensive phenotype profiling was done to assess the expression of 17 markers, CD24, CD29, CD34, CD44, CD45, CD73, CD90, CD105, CD117, CD133, CD146, CD200, CD271, ABCG2, HLA-ABC, HLA-DR, and STRO-1 that have been found relevant in previous studies.32–34 The distribution of six positive markers is illustrated for the ASC23 line in Figure 2A. Some variability is apparent between the three growth conditions; however, when taking into account also the other cell lines, no statistically significant patterns were observed at P9 (Fig. 2B). The spread of marker representation across the investigated cell lines can also be appreciated from the size of standard errors of mean.
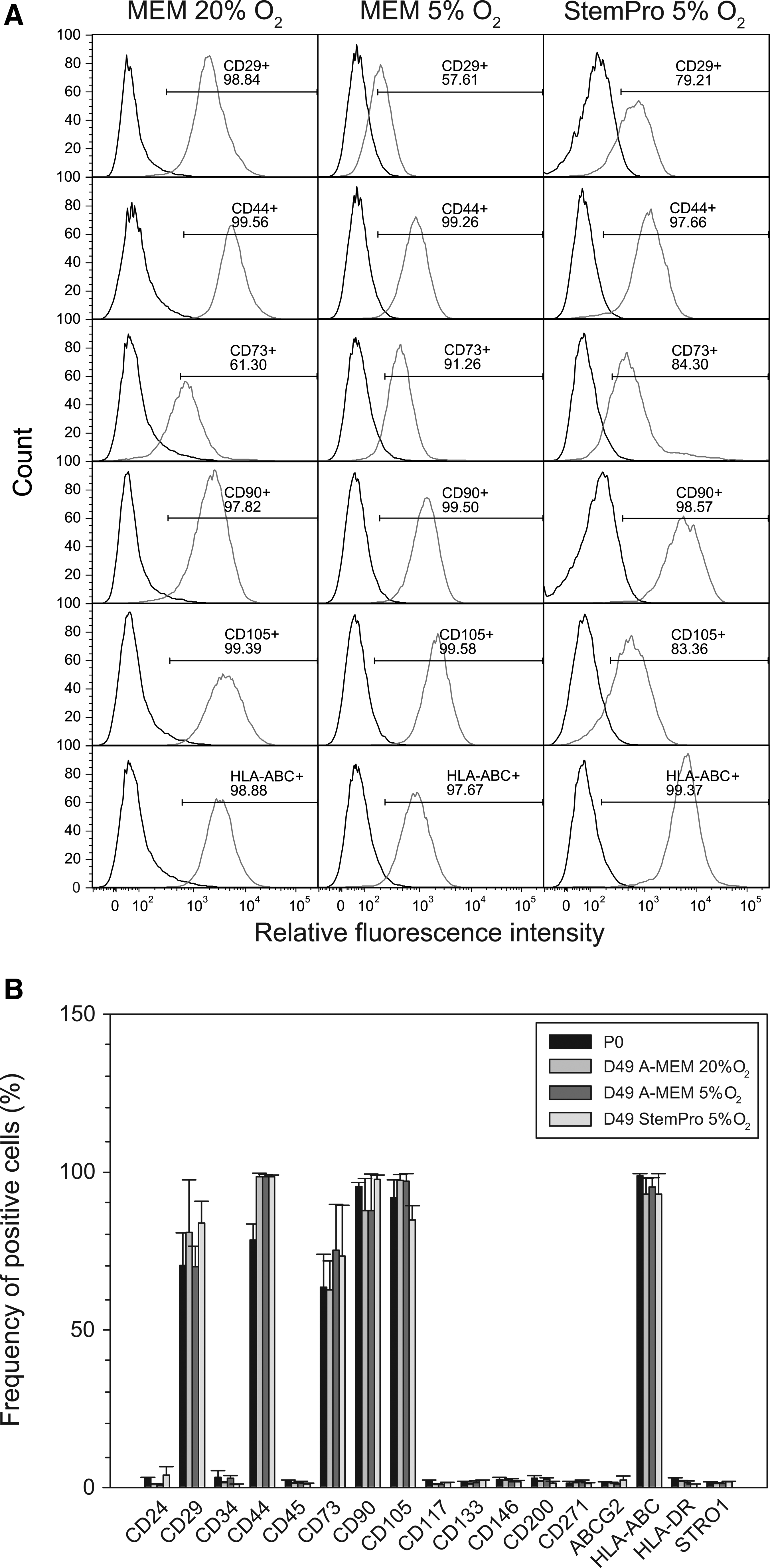
Immunophenotypic profile of human ASCs during long-term expansion in different growth media and oxygen concentrations.
When looking at the temporal changes over time, a difference between the initial (P0) and final (P9) phenotype profiles under the different growth conditions was prominent for CD44. Despite the increasing proportion of positive cells from 78.5% to 98.5% on average, this change was not statistically significant. Similarly, no significant changes were observed for the remaining positive markers during the course of the 49-day expansion.
Maintenance of differentiation potential in ASCs expanded during long-term culture
The specific adipogenic, osteogenic, and chondrogenic multipotent differentiation potential of expanded ASCs was determined by cytochemical approaches to detect lipid synthesis, calcium deposition, and production of glycosaminoglycan matrix, respectively (Fig. 3A). Adipogenic conditions induced a marked accumulation of lipid droplets in monolayers of cells from all three growth conditions as revealed by staining with oil red O. Similarly, in the osteogenic conditions, the visualization with alizarin red identified a comparable amount of calcified matrix in cultures derived from all three growth conditions. In the chondrogenic conditions, images of the whole micropellets illustrate the process of compaction and solidification as the newly produced extracellular matrix embedded the cellular component in samples from all growth conditions. With respect to the size of differentiated pellets, the cells being expanded in A-MEM+5% O2 supported the biggest tissue volumes, when normalized to that of control cultures. The histochemical analysis further confirmed the macroscopic pattern and revealed the structural features of the cartilage tissue. The cells from all three growth paradigms supported production of a rich cartilage-specific extracellular matrix based on acidic glycosaminoglycans, as indicated by a deep alcian blue staining.

Evaluation of differentiation potential of ASCs maintained in different growth media and oxygen concentrations during a long-term expansion.
In addition to phenotypic characterization of differentiating cultures by cyto- and histochemistry, analysis of transcriptional expression of factors central to the particular differentiation pathway was determined by real-time reverse transcriptase–PCR. The transcription factors PPAR-γ2, Cbfa1, and Sox9, which are involved in adipo-, osteo-, and chodrogenic specifications, respectively, were investigated (Fig. 3B). The gene expression regulation under the tested hypoxic conditions is presented in relation to the normoxic control conditions. The normalized values revealed similar levels of transcriptional activation when A-MEM and StemPro were compared. Only Sox9 in A-MEM under hypoxia was significantly upregulated with respect to the control condition, confirming previous observations from other cell systems that moderate levels of hypoxia promote chondrogenic differentiation.35–38 There was also an indication of a pattern that in hypoxia the analyzed factors were less expressed in StemPro than in A-MEM; however, this difference could not be supported at a statistically significant level.
Discussion
From a clinical application perspective, it is desirable that the stem cells retain their stemness and, moreover, are efficiently expanded in the absence of xenogeneic components that may represent the risk of adverse immune reactivity and potential transmission of infectious agents. Commercial products based on fully- or semi-defined supplements to replace FCS for the growth of MSCs are available; however, the performance of these products does not always compare favorably with that of FCS. 39 The StemPro growth system has been tested previously, and the results demonstrate that it well satisfies the demands for a high-performance scale-up of fully functional mesenchymal precursors.11,12,40 Importantly, in addition to biochemical signaling, numerous physical factors that include, for example, surface elasticity and roughness,41,42 mechanical force, 43 and temperature changes,44–46 have been shown to influence stem cell responses. Since oxygen has especially been documented to support the maintenance of stem cell differentiation potential and accelerated growth, it was appealing to further optimize the StemPro growth conditions within the context of a hypoxic environment. The experimental evidence, however, is conflicting as to the optimal level of oxygen concentration, and it appears that different types of stem cells may require different levels of hypoxia. For example, the self-renewal of human ESCs is best maintained in 5% oxygen,13,14 while that of limbal stem cells at 2%. 16 With respect to ASCs, our own comparative studies identified 5% oxygen in culture atmosphere as optimal to support growth,22–24 although it is possible that in certain settings, even lower oxygen concentrations may prove useful.47,48 The somewhat disparate experimental evidence clearly indicates that better standardization of hypoxic experiments together with identification of possible pitfalls is necessary before consensus on a widely accepted value can be reached.
It is generally accepted that during the course of in vitro expansion, the population of primary stem cells inevitably undergoes the process of senescence that may affect the differentiation potential or other regenerative capacities.49–52 It is unfortunate that many studies define the expansion period only in terms of passages, which precludes direct comparison. The only objective reference with respect to which the age of cultures should be expressed is the number of Nd. Just a minor change in the Td, as for example from 2 to 1.9 days, results in a 15% higher proliferation rate over a single passage (7 days), which over further 5 passages translates in a twofold increase in the total number of cells. Thus, under extremely high proliferation rates, such as in the described expansion of ASCs in StemPro under hypoxia, the cultures rapidly acquire a larger number of Nd, and the effect of senescence may come into effect earlier than under standard growth conditions. This may provide at least a partial explanation for the observed two-phase proliferation profile in the StemPro and hypoxia condition.
The initial population of cells isolated from fat stroma is rather heterogenous and appears to contain several types of progenitors.53–55 Upon selection through adherence to a tissue culture-treated plastic surface, the consensus phenotypic profile of ASCs has been proposed to include the presence of CD44, CD73, CD90, CD105, CD146, and HLA-ABC and a lack of CD14, CD31, CD34, CD45, and HLA-DR. 32 Other markers, such as CD24, CD200, or CD271, have been proposed to complement the basic repertoire,33,34 but further work is necessary to establish their significance. In this investigation, we confirmed the canonical ASC immunophenotype and further demonstrated that it remained stable in the course of extensive expansion. In the context of previously reported temporal changes during in vitro culturing of ASCs, 49 the only conspicuous feature we observed, was a trend toward higher CD44 expression. In the face of insufficient statistical support, however, additional experimental data appear necessary to confirm this observation.
In addition to the expression of surface determinants, the maintenance of multipotency, as most frequently assayed through adipo-, osteo-, and chondrogenic differentiation, is another critical surrogate marker for the regenerative capacity of ASCs. It is interesting that historical experimental data lend strong support to the notion that the regenerative capacity of ASCs is fairly robust and even during extended in vitro expansion remains well maintained.47,56–58 The current investigation provides further evidence that fully corroborates the previous findings on the maintenance of ASC multipotency. Taken together, our protocol proposes, through integration of xenogeneic-free and hypoxic conditions, a new modality that can be beneficially implemented in the future regenerative and tissue engineering approaches based on ASCs or other MSCs.
Footnotes
Acknowledgments
The authors wish to thank H. S. Møller for technical assistance. The financial support from the Carlsberg and the John and Birthe Meyer foundations is highly appreciated.
Disclosure Statement
No competing financial interests exist.