
Abstract
Adequate cellular in-growth into biomaterials is one of the fundamental requirements of scaffolds used in regenerative medicine. Type I collagen is the most commonly used material for soft tissue engineering, because it is nonimmunogenic and a highly porous network for cellular support can be produced. However, in general, adequate cell in-growth and cell seeding has been suboptimal. In this study we prepared collagen scaffolds of different collagen densities and investigated the cellular distribution. We also prepared a hybrid polymer–collagen scaffold to achieve an optimal cellular distribution as well as sufficient mechanical strength. Collagen scaffolds [ranging from 0.3% to 0.8% (w/v)] with and without a mechanically stable polymer knitting [poly-caprolactone (PCL)] were prepared. The porous structure of collagen scaffolds was characterized using scanning electron microscopy and hematoxylin-eosin staining. The mechanical strength of hybrid scaffolds (collagen with or without PCL) was determined using tensile strength analysis. Cellular in-growth and interconnectivity were evaluated using fluorescent bead distribution and human bladder smooth muscle cells and human urothelium seeding. The lower density collagen scaffolds showed remarkably deeper cellular penetration and by combining it with PCL knitting the tensile strength was enhanced. This study indicated that a hybrid scaffold prepared from 0.4% collagen strengthened with knitting achieved the best cellular distribution.
Introduction
Type I collagen is the most commonly used biomaterial for soft tissue. It is highly abundant in the human body, and it is biocompatible, 3 low immunogenic, structurally supports cell growth, 4 and provides a natural environment for cell growth, differentiation, and wound repair. These excellent qualities have made collagen a preferred material for regenerative medicine purposes for more than a decade. 5 In general, collagen scaffolds are produced by lyophilization of collagen solutions followed by cross-linking to achieve better mechanical characteristics and to lower antigenicity. 6 In earlier studies using collagen scaffolds prepared from 0.8% (w/v) type I collagen, we noticed poor cellular in-growth into the scaffolds. 7 Thus, despite the highly porous character of collagen scaffolds, cell in-growth must be improved to achieve a better outcome.
Many studies have been performed to enhance the cell distribution inside scaffolds: static seeding and dynamic seeding. Static seeding, which is the most common technique, includes surface seeding 8 and injection seeding into the scaffold. 9 Dynamic seeding includes spinner flask seeding, 10 agitation in a bioreactor,11,12 and perfusion. 13 Dynamic seeding is effective and the cell proliferation might be improved by physical agitation. 14 Another approach is to improve the characteristics of the construct. We hypothesized that scaffold interconnectivity, leading to improved cell penetration, can be enhanced by decreasing the collagen content. Nevertheless, by lowering the collagen content, the mechanical properties of the collagen diminish and handling becomes impossible (decreased fixation). Combining the scaffold with other materials like polymers may alleviate this problem. Many clinically applicable materials are available for this purpose, for example, poly-glycolic acid, poly-lactic acid, and poly-caprolactone (PCL). In this study we introduced a new hybrid collagen–polymer scaffold with optimal cellular distribution and mechanical strength. As an example for a multilayered organ, we used the bladder, and investigated the cell distribution in the novel construct and enhance the engineering of this organ.
Materials and Methods
Collagen suspension
Collagen scaffolds were prepared from purified type I collagen as described by Pieper et al. 15 Insoluble type I collagen fibrils were purified from pulverized bovine Achilles tendon. The purification consisted of washings with diluted acetic acid, aqueous NaCl solutions, urea, acetone, and demineralized water. Collagen [0.3%, 0.4%, 0.5%, 0.6%, 0.7%, and 0.8% (w/v)] was suspended in 0.25 M acetic acid and swollen overnight at 4°C. Then, the collagen was homogenized on ice using a Potter-Elvehjem homogenizer. Air bubbles were removed by centrifugation (10 min, 250 g at 4°C) and suspensions were poured into six-well plates.
Poly-caprolactone
PCL was purchased from Sigma-Aldrich. The molecular weight is 80,000 g/mol. Knittings were prepared from PCL threads, 16 which were knitted into sheets according to the stockinet structure using knitting machinery (Lawson Hemphil).
Hybrid scaffolds
For the hybrid scaffolds the bovine collagen solution was poured on top of the man-made PCL knittings. Air bubbles were removed using an additional centrifugation step (10 min, 250 g at 4°C). The scaffolds were subsequently frozen and lyophilized in a Zirbus Sublimator 500II. Carbodiimide cross-linking was performed to cross-link the scaffolds. 17 In brief, lyophilized scaffolds were preincubated in 2-morpholinoethane sulfonic acid buffer in 40% ethanol (pH 5) overnight at 4°C. Cross-linking was performed with 33 mM EDC [N-ethyl-3-(3-demethylaminopropyl)carbodiimide] and 6 mM NHS (N-hydroxysuccimide) in 50 mM 2-morpholinoethane sulfonic acid (pH 5.0) containing 40% ethanol for 4 h at 20°C. The scaffolds were then washed in 0.1 M Na2HPO4, 1 M NaCl, 2 M NaCl, and water followed by 70% ethanol and stored at −20°C. Before use, the scaffolds were washed in 70% ethanol (4×1 h and 1×overnight), followed by washings in sterile phosphate-buffered saline (PBS; pH 7.4, 4×1 h, 1×overnight), and followed by an overnight incubation in the culture medium.
Scanning electron microscopy
The structure of hybrid collagen–PCL scaffolds was examined by a scanning electron microscope (SEM) (JSM-6310; JEOL). Scaffolds were fixed with 2% (v/v) gluteraldehyde, and then dehydrated by graded series ethanol (30%–100%) and finally dried using the critical point dryer (Polaron; Quorum Technologies). Samples were sputtered with gold for 60 s by Scancoat Six SEM Sputter Coater (Temescal) before being studied.
Determination of cross-linking
Samples were incubated with 4% (w/v) NaHCO3 in water for 30 min at 21°C, and 0.5% (w/v) 2,4,6-trinitrobenzene sulfonic acid solution was added and the samples were incubated for 2 h at 40°C. The degree of cross-linking was determined by the change of free amine groups by measuring the absorbance of resultant trinitrophenylated amine complex at 420 nm (Biorad).
Determination of water retention
Samples were incubated with water for 24 h at 21°C on the shaker. The weight of wet scaffolds was weighed after removing the excess water with blotting paper. The weight of dry scaffolds was determined after freezing and lyophilization (VirTis Sentry) overnight. At each percentage of collagen scaffolds, six samples were tested. The water retention was calculated according to the formula:
Where Wwet is the wet weight after blotting and Wdry is the weight after drying.
Ultimate tensile strength
Samples were preincubated in 0.9% NaCl, cut into strips, and placed between clamps in a bioreactor (Bose Electroforce Biodynamic Bioreactor). The measurements were recorded and controlled by the WinTest v4.1. The elongation speed was set at 20 mm/min and forces were recorded with a 22 N load cell. The distance between clamps was 10 mm for collagen samples and 5 mm for PCL plus collagen samples. The valley and peak distance was 12 mm. The measurements were repeated at least 4 times for each scaffold of a particular collagen density. Measurement was valid when a sample broke in the middle.
Distribution of fluorescent beads
The scaffolds were washed five times in PBS before bead seeding. The fluorescent beads (10-μm diameter, 488 nm, Flow Check; Beckman Coulter) were washed using 5% (w/v) bovine serum albumin (BSA; Sigma Aldrich) in PBS. The fluorescent beads were seeded at a density of 2×106 beads/cm2 scaffold and incubated overnight under continuous shaking at 21°C. The bead-seeded scaffolds were collected and incubated for 20 min in Tissue-Tek (Sakura Finetek Europe B.V.) and snap-frozen. Cryostat sections (5 μm) were collected on gelatin-coated slides [0.5% gelatin (w/v), 0.05% potassium chrome (III) sulfate (w/v), and demi-water], fixed with 100% methanol, and air dried for 1 h at 21°C.
Cell culture
Human bladder smooth muscle cells (HBSMCs) were purchased from Sciencell (Carlsbad). HBSMCs were cultured in smooth muscle cell medium (SMCM, Cat. 1101; Sciencell) with 2% (v/v) fetal bovine serum, 1% (v/v) smooth muscle cell growth supplement, and penicillin (100 iU/mL)–streptomycin (100 μg/mL). Human urothelial cells (ScaBER, derived from squamous cell carcinoma of human bladder) were cultured in RPMI-1640 medium (Cat. 21870; Invitrogen) with 10% (v/v) fetal calf serum, without glutamin, and penicillin (100 iU/mL)–streptomycin (100 μg/mL). Cells were cultured and expanded at 37°C and 5% CO2 in humid atmosphere air.
Cell seeding
Static seeding of HBSMCs on the scaffold was performed using 2.6×106 cells/cm2. Cells were cultured for 14 days and medium was replaced every 2–3 days. Thereafter, the scaffolds were incubated in RPMI-1640 medium overnight followed by an urothelial cell seeding step. ScaBER cells (6.6×106 cells/cm2) were seeded, and scaffolds were incubated for an additional 7 days.
Immunohistochemistry
Cell-seeded scaffolds were fixed in 4% (v/v) formalin in PBS overnight and embedded in paraffin. Four-micrometer sections were cut (Microm), deparaffinized in xylene, and rehydrated using a graded series of ethanol, and stained with hematoxylin-eosin (H&E).
For immunohistochemistry analysis, 5-μm cryostat sections of slides were fixed in methanol for 10 min at −20°C and air dried for 1 h at 21°C. Sections were blocked with 10% (v/v) normal goat serum in PBS for 1 h. Then, sections were incubated with various primary antibodies, such as rabbit anti-bovine type I collagen (Millipore; 10 μg/mL), mouse anti-human RGE53 (MUbio BV; 10 μg/mL), rabbit anti-human vimentin (Pathology, UMC St. Radboud Nijmegen; 1:200), and mouse anti-human CD90 (Dianova GmbH; 40 μg/mL), in blocking buffer for 1 h at 21°C in a wet chamber. The primary antibodies were developed with goat anti-rabbit Alexa-488 (Invitrogen; 10 μg/mL) or goat anti-mouse Alexa 594 (Invitrogen; 10 μg/mL). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole in Prolong gold mount medium (Invitrogen). The samples were evaluated by fluorescence microscopy (LEICA DC 300F).
Results
Characterization of the collagen scaffolds
SEM and H&E and images of the different collagen scaffolds are shown in Figures 1 and 2. The scaffolds had the typical honeycomb morphology. 18 The pore size of all collagen scaffolds ranged between 75 μm and 150 μm, and the degree of cross-linking was ∼43%, 18 regardless of collagen density. The water retention capacity of the scaffolds was highly similar after 24 h of incubation. In general, different density scaffolds revealed more connections between pores in the lower density scaffolds as compared with the higher density scaffolds (Fig. 1).

Microscopic characterization of the cross-sectioned collagen scaffolds. Scanning electron microscopy
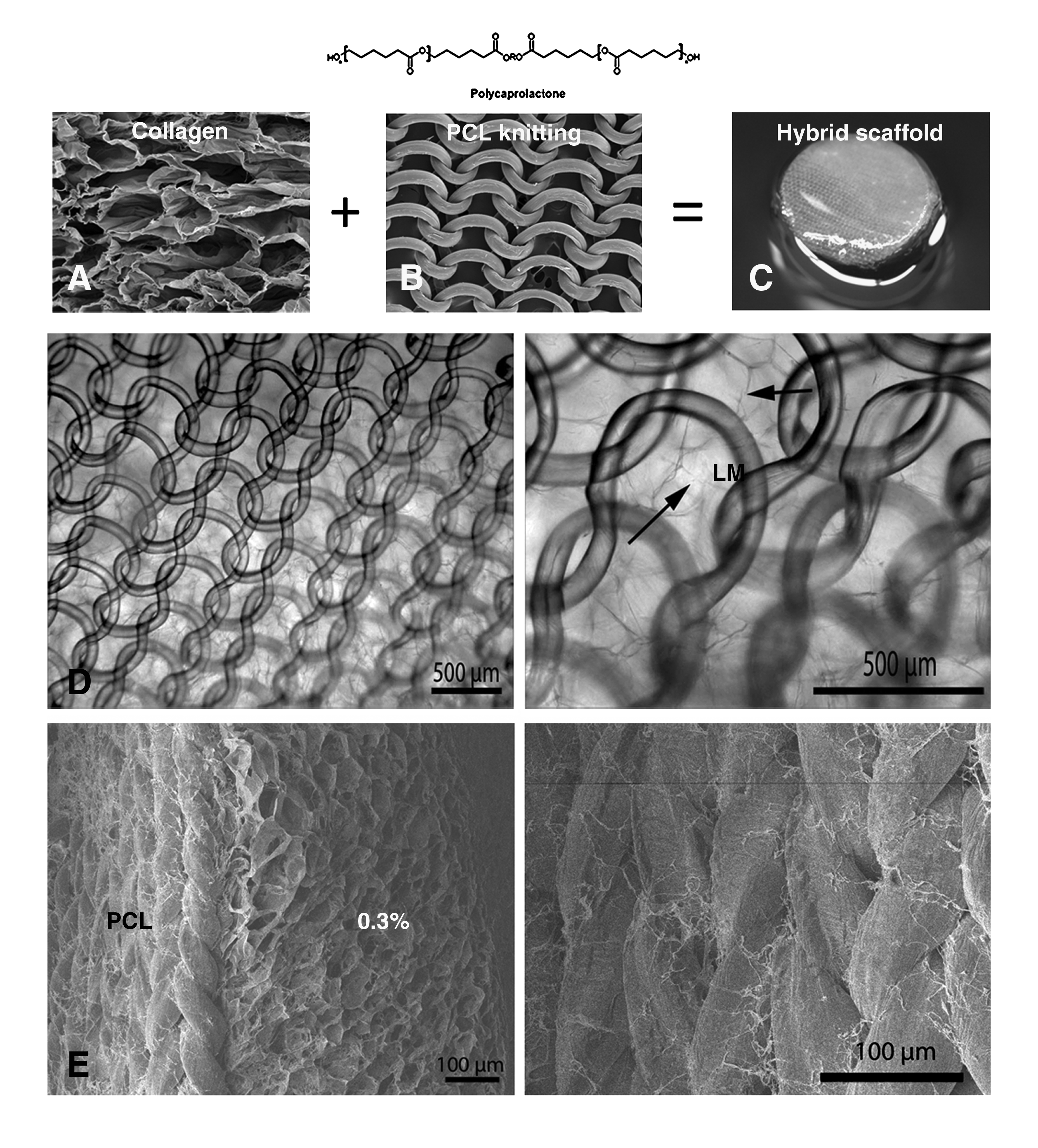
Schematic overview of preparation hybrid scaffold. Type I collagen
The hybrid scaffolds were prepared by pouring the collagen solutions on top of the PCL knitting. Despite the fact that no additional measures were taken, the collagen and PCL appeared to be adequately connected (see Fig. 2). The woven structure of the PCL knitting may have aided in an adequate collagen distribution.
Tensile strength experiments were performed to determine the mechanical properties of the scaffolds with and without PLC knitting (Fig. 3a). The strength of scaffolds increased slightly as the density of the collagen increased (R2=0.97; p=0.0008). Further, hybrid scaffolds were much stronger compared with scaffolds composed of collagen alone (Fig. 3b, c). Even at the highest tensile force applied, the PCL–collagen scaffold did not break but deformed; the collagen layers broke and slid from the PCL and the morphology of the PCL knitting was irreversibly changed.
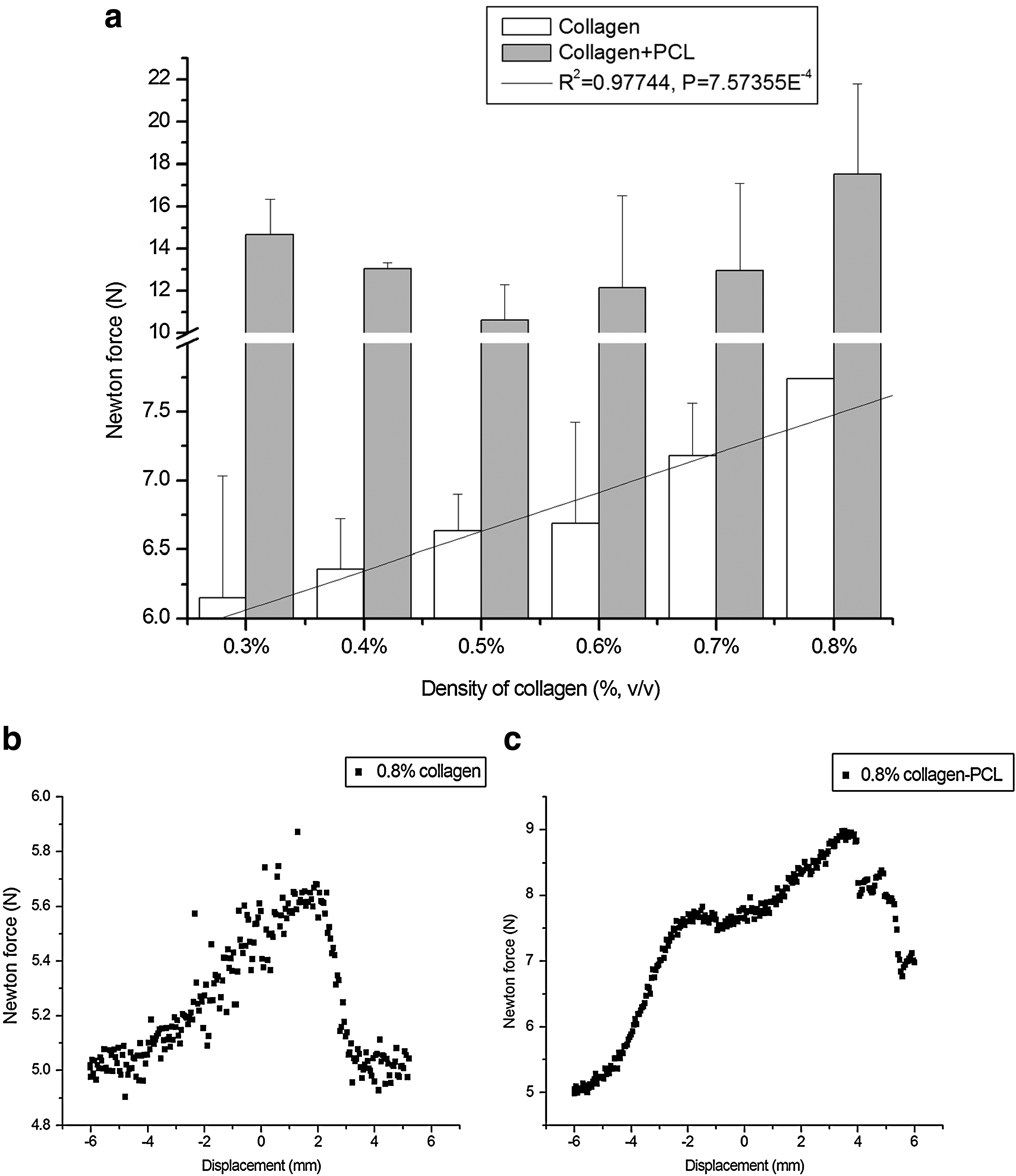
Tensile test of collagen scaffold with and without PCL
To further investigate the influence of the collagen concentration on the interconnectivity of the resultant different scaffolds, the distribution of fluorescent beads was investigated. To mimic cell penetration, 10-μm-diameter beads were used, comparable with the size of animal eukaryotic cells. The distribution of the beads was highly dependent on the collagen density; at lower collagen densities the beads were more evenly distributed and penetrated deeper into the scaffold (see Fig. 4). Beads presented throughout the 0.3% and 0.4% scaffold and a less homogeneous distribution was noted for the 0.5% and 0.6% scaffold, whereas the fluorescent beads were mainly on the surface of the 0.7% and 0.8% collagen scaffolds.
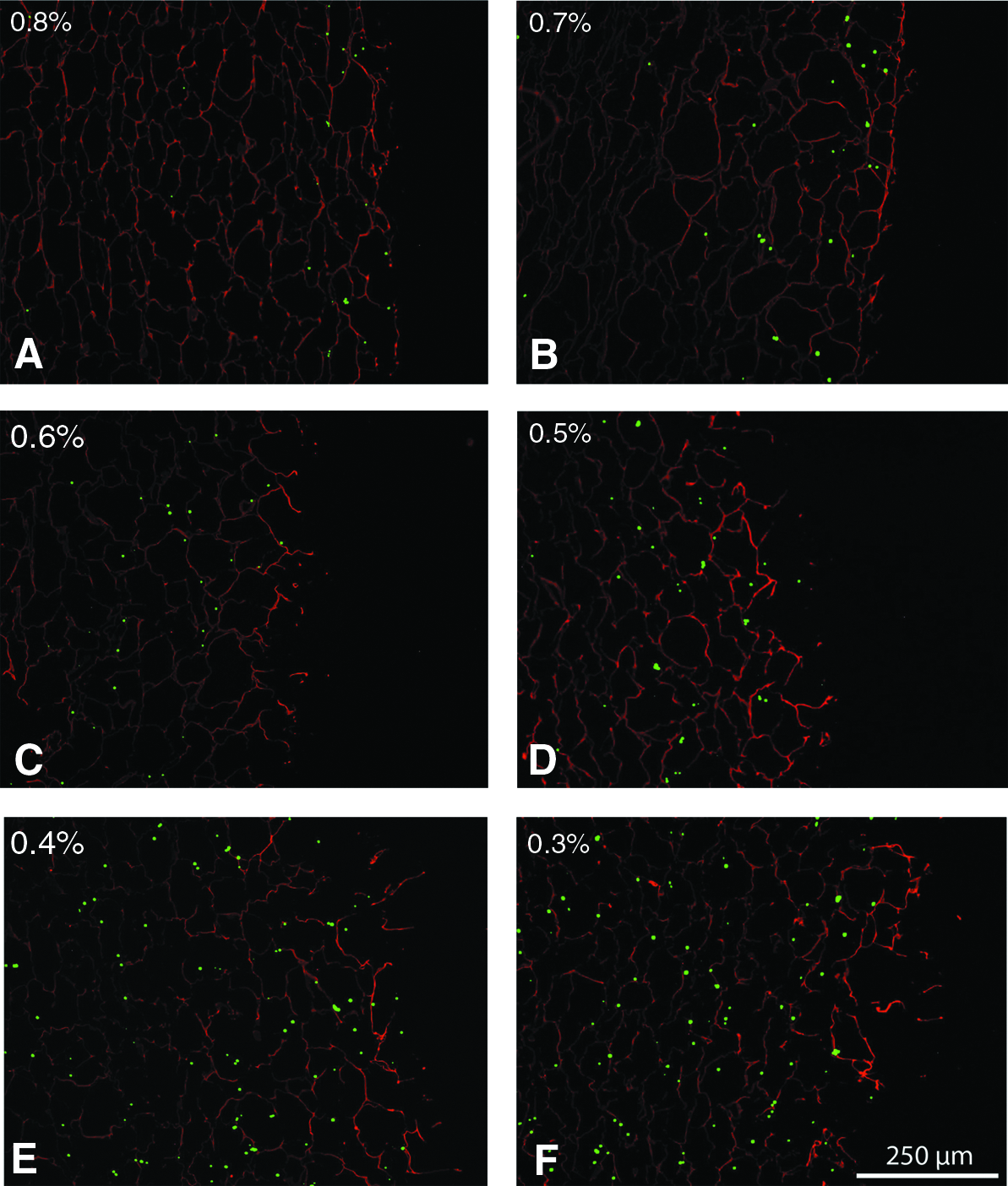
Bead distribution in collagen scaffolds (0.8%–0.3%,
Cell distribution
To mimic multilayered cell growth, human bladder smooth muscle cells (HBSMCs) were seeded onto the scaffolds followed by urothelial (ScaBER) cells. HBSMCs were allowed to grow for 14 days, in an effort to generate scaffolds mimicking the urinary bladder wall, the natural stratum for urothelial cells. HBSMCs were visualized with an anti-human smooth muscle cell marker (CD-90 and vimentin). The distribution of HBSMCs inside the scaffolds was more homogeneous in the lower collagen density scaffolds (0.3% and 0.4% scaffolds) where they almost completely covered the depth of the scaffolds (see Figs. 5 and 6). HBSMCs in-growth of scaffolds of higher collagen content was less extensive, and less dispersed. Inclusion of PCL did not influence the cell distribution.
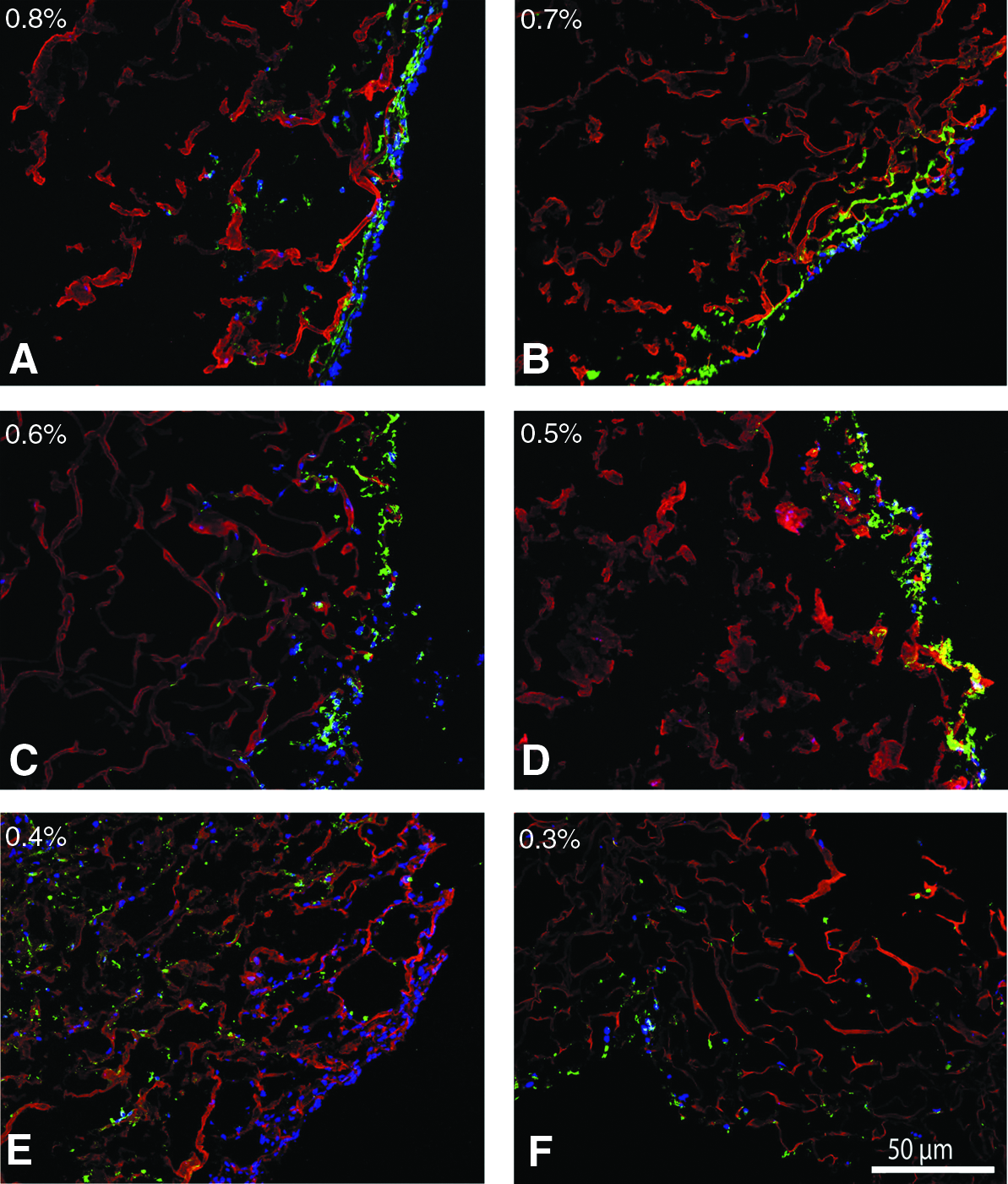
Immunofluorescence (IF) analysis of cell distribution in different densities (from 0.8% to 0.3%,
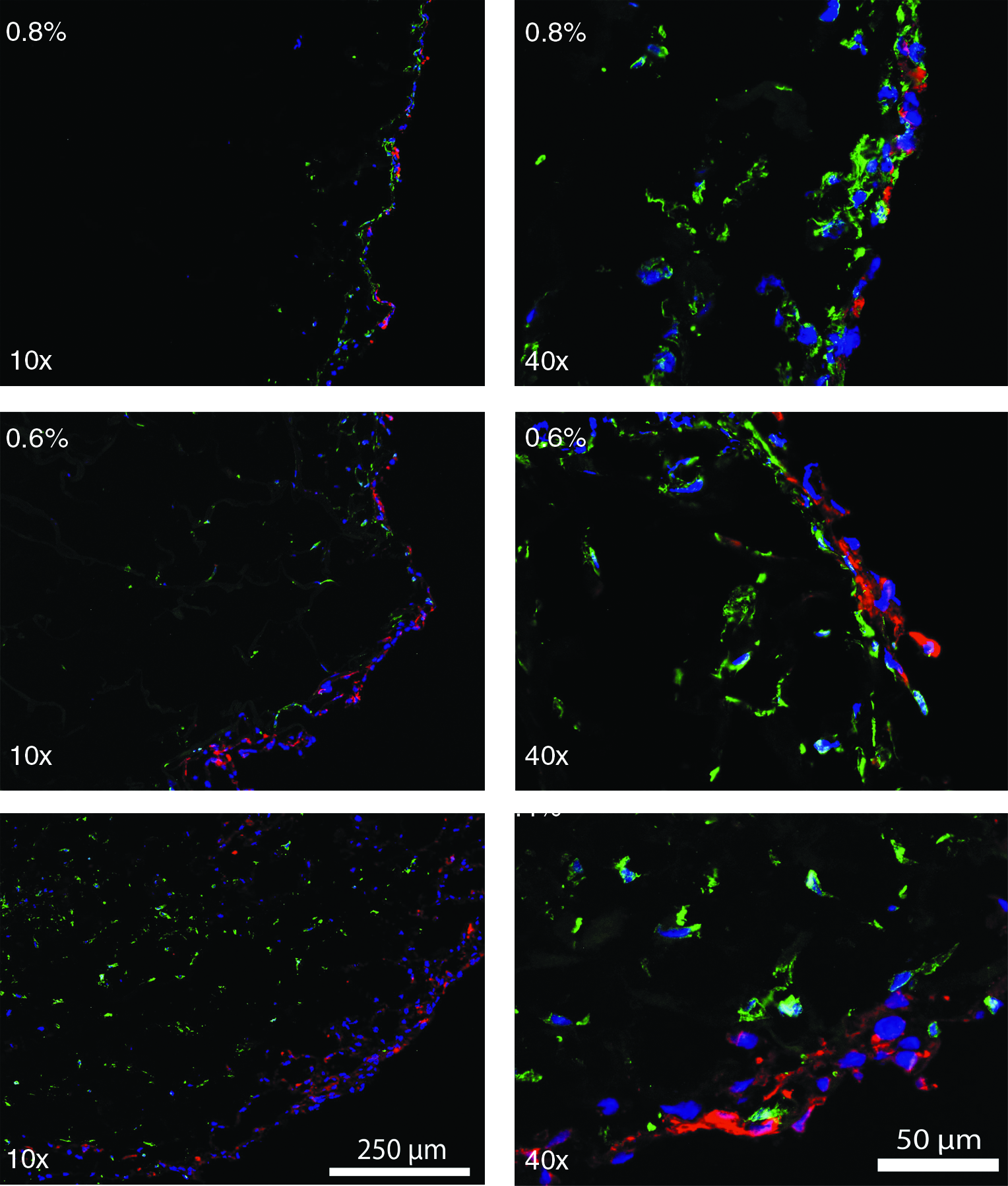
IF double staining of HBSMCs and ScaBER in different densities (0.8%, 0.6%, and 0.4%) of collagen scaffold. HBSMCs were developed by Alexa 488 (green), ScaBER was developed by Alexa 594 (red), and all nuclei were counterstained by DAPI (blue). HBSMCs and ScaBER in scaffolds were imaged by fluorescence microscopy (10×and 40×). HBSMCs are located under the ScaBER cells as natural stratum for urothelial cells. ScaBER cells are always on top of the scaffolds regardless of the collagen density. More HBSMCs are distributed in the low-density scaffolds.
Seeding of bladder cells resulted in urothelial cell growth restricted to the scaffold surface, mimicking the build-up of normal bladder tissue. Even after 3 weeks of culture, the scaffolds supported the cells and did not lose integrity, regardless of collagen content. The distribution of the bladder cells was visualized with an anti-human cytokeratin marker (RGE-53) (see Fig. 6). Comparison of cell markers of cells grown under standard cell culture conditions did not reveal any difference with cells grown on scaffolds.
Discussion
For soft tissue engineering, adequate cell colonization is crucial. Failure of cellular in-growth can lead to scar formation, and foreign body reactions leading to an undesired end result. Collagen has been applied as biomaterial for soft tissue engineering for many years, but the issue of adequate cell in-growth is still unresolved. 7 Addition of migrational and differentiating cues may alleviate this problem, but thus far success has been limited. 19 One of the parameters that influence cell distribution is scaffold porosity and interconnectivity. To improve the cell distribution in honeycomb collagen scaffolds and to optimize their use for soft tissue engineering, we prepared 0.3% to 0.8% (w/v) collagen scaffolds and included a rigid knitting to improve the mechanical and handling characteristics. The porosity of all scaffolds was similar, but lowering the collagen content resulted in higher interconnectivity and improved cellular in-growth. Cells remained on the surface of the higher density collagen scaffolds, but almost completely invaded the scaffolds of the lowest density, leading to a scaffold almost homogeneously occupied with HBSMCs. Thus, the functionality of the collagen scaffolds with regard to the cellular distribution was improved by lowering the collagen density.
The low-density collagen scaffolds were very fragile, which made them unsuitable for in vivo use. To improve the mechanical strength polymer knitting was incorporated. This resulted in a hybrid scaffold with significantly increased mechanical and functional characteristics; cell in-growth was greatly enhanced due to the lower collagen content whereas the hybrid scaffold could be handled easily due to the incorporation of the polymer. The polymer used, PCL, is frequently used as biomaterial. The degradation time of PCL is ∼6 to 9 months in vivo, very suitable for implantable systems where scaffolds must provide temporary mechanical support for a longer period until the regenerated tissue can support mechanical loads. 20 Incorporation of supportive materials other than PCL—for instance, copolymers of lactic acid, glycolic acid, or trimethylene carbonate by which the degradation time can be tailored19,21–23—should greatly enhance the implementation possibilities for soft tissues. Similar to our scaffolds, PLGA- and PLLA-enforced hybrid scaffolds have been prepared for cartilage regeneration.24,25 In these hybrids a favorable cell distribution was observed, most likely related to the unidirectional orientation of the pores. In our honeycomb-structured scaffolds that are needed to regenerate complex, multilayered tissues composed of multiple cell types, we needed to decrease the collagen content of the hybrid scaffolds to achieve a favorable cell distribution for soft tissue regeneration. Through small changes in collagen content we could control the cellular distribution.
To further quantify the effect of lowering the collagen content, we studied the distribution of 10-μm-diameter fluorescent beads. The distribution of the beads was identical to the distribution of cells, with better bead penetration at the lower collagen densities. This again demonstrates that the interconnectivity increased by lowering the collagen content. Obviously, the bead distribution does not truly reflect the cell distribution after cell seeding, as other factors such as cell surface influence this distribution. The obvious advantages of the beads are the rapidity with which the results are obtained; the consistency, because variability due to the use of different cells is lacking; and the objectivity, with the possibility to vary bead size to judge interconnectivity and measure bead penetration. 26 Many studies have applied different seeding methods to improve the cellular distribution in honeycomb-like scaffolds. Different procedures of dynamic cell seeding have been attempted, including centrifugation, 27 rotation, 28 magnetic field, 29 and vacuum, 30 in a bioreactor. Nevertheless, almost invariably, the cell distribution was limited to the scaffold surface with little spreading throughout the scaffold. Coating of PET fabrics with 0.3% collagen (without cross-linking) demonstrated an even cellular distribution throughout 1.5-mm scaffolds 28 similar to our observations with 5-mm-thick scaffolds prepared from 0.3% collagen. Our results suggest that the efficiency of cell colonization largely depends on the collagen density of the scaffold. Most likely, the higher interconnectivity in the scaffold facilitates cellular migration since collagen degradation is not necessary to colonize adjacent pores of the scaffold. Since cellular in-growth appears to be greatly dependent on the pore interconnectivity, it is not unexpected that static seeding at higher cell density did not lead to better and more homogeneous cell growth.
Lowering the collagen content may be more beneficial in achieving a homogeneous cell distribution than active cellular seeding. However, improved cell in-growth in vivo in lower collagen content scaffolds remains to be studied. In addition, active cell seeding in this low collagen content, polymer-supported scaffolds may lead to a more homogeneous cell distribution.
Conclusion
In summary, the functionality of collagen scaffolds can be greatly improved by lowering the collagen content and including polymer knitting, leading to a versatile scaffold with new opportunities for soft tissue engineering.
Footnotes
Acknowledgments
The research leading to these results has received funding from the European Community's Seventh Framework Programme (MultiTERM, grant agreement No. 238551) and European Commission (EuroSTEC, 6th Framework, LSHC-CT-2006-037409).
Disclosure Statement
No competing financial interests exist.