
Abstract
Mesenchymal stem cells (MSCs) obtained from human bone marrow are pluripotent and have been expanded and differentiated into several kinds of mesodermal tissue in vitro. To create bioartificial tissues and organs for implantation, it is necessary to induce proliferation in such cells. In this study, a radial-flow bioreactor (RFB) was used to induce three-dimensional (3D) expansion of human MSCs (hMSCs) on a large scaffold. The effect of this expansion on cellular characteristics was investigated. To produce precultured sheets, the hMSCs were first seeded onto type 1 collagen sheets and incubated for 12 h, after which they were placed in the RFB for fabrication of scaffolds. The culture medium was circulated at 3 mL/min, and the cells were dynamically cultured for 1 week at 37°C. As a control, static cultivation in a culture dish was also carried out. Cellular expansion and characteristics were analyzed. Alkaline phosphatase (ALP) activity in the hMSCs was also investigated after dynamic culture in an osteogenesis induction medium to explore their potential for osteogenic differentiation. At 1 week of dynamic cultivation, a >60% increase was observed in a number of cells together with a uniform distribution throughout the scaffolds compared with under static conditions; no change in hMSC markers was observed. The hMSCs retained the ability for osteogenic differentiation after culture in the RFB. The present results indicate that 3D dynamic culture in an RFB enables uniform expansion of hMSCs with no change in cellular characteristics, suggesting the usefulness of this technique in tissue engineering.
Introduction
MSCs harvested this way could provide a valuable resource as long as they retained all of their normal potential for self-renewal and multilineal differentiation. 4 Human MSCs (hMSCs) are pluripotent bone marrow cells, and have been expanded and differentiated into several kinds of mesodermal tissue, including cartilage, fat, and bone, in vitro. 5 Induced pluripotent stem cells and embryo stem cells, in particular, have great potential for the future of tissue engineering, as they would offer infinite capacity for proliferation and differentiation. However, many problems remain to be resolved with regard to these techniques, including ethics issues and the problem of tumorigenic transformation. Therefore, it is too early for a clinical application, as yet. In contrast, somatic stem cells, as represented by MSCs, have a good track record in basic research; their safety has been confirmed, and some clinical research employing such cells is already underway.
This increased interest in tissue engineering has led to the development of various types of equipment for the construction of 3D cultured tissues. Bioreactors, of which spinner flasks 6 and rotating vessels7,8 are two examples, provide the conditions necessary for the maintenance and promotion of tissue culture. In this type of bioreactor, the scaffolds are fixed or allowed to float, with the culture medium being changed at specific intervals. The perfusion bioreactor is another example, in which the scaffolds are fixed, and the medium is continuously circulated throughout the chambers. The radial-flow bioreactor (RFB) is one such bioreactor. This type has shown the ability to maintain an even cell culture environment by radial provision of the medium,9–12 allowing comparatively larger tissues to be constructed. The RFB offers highly functional 3D cultivation. To allow even distribution of oxygen, the medium is pumped to the center of the chamber from the periphery under low shear stress. High-density 3D culture of HepG2 cells was successfully carried out in the construction of artificial liver.9–11
Arano et al. reported culture of mouse osteoblastic cells (MC3T3-E1) on a collagen scaffold using an RFB. 12 Since MC3T3-E1 are immortalized, it is easy to use them for study. However, it would be inappropriate to use such cells for tissue engineering in humans.
Therefore, this study aimed to investigate whether an RFB enabled uniform expansion of hMSCs on a 3D scaffold with same method as a previous study that used the osteoblastic cells. 12 In addition, the characteristics of hMSCs after 3D culture were examined.
Materials and Methods
Figure 1 shows a summary of the study protocol.
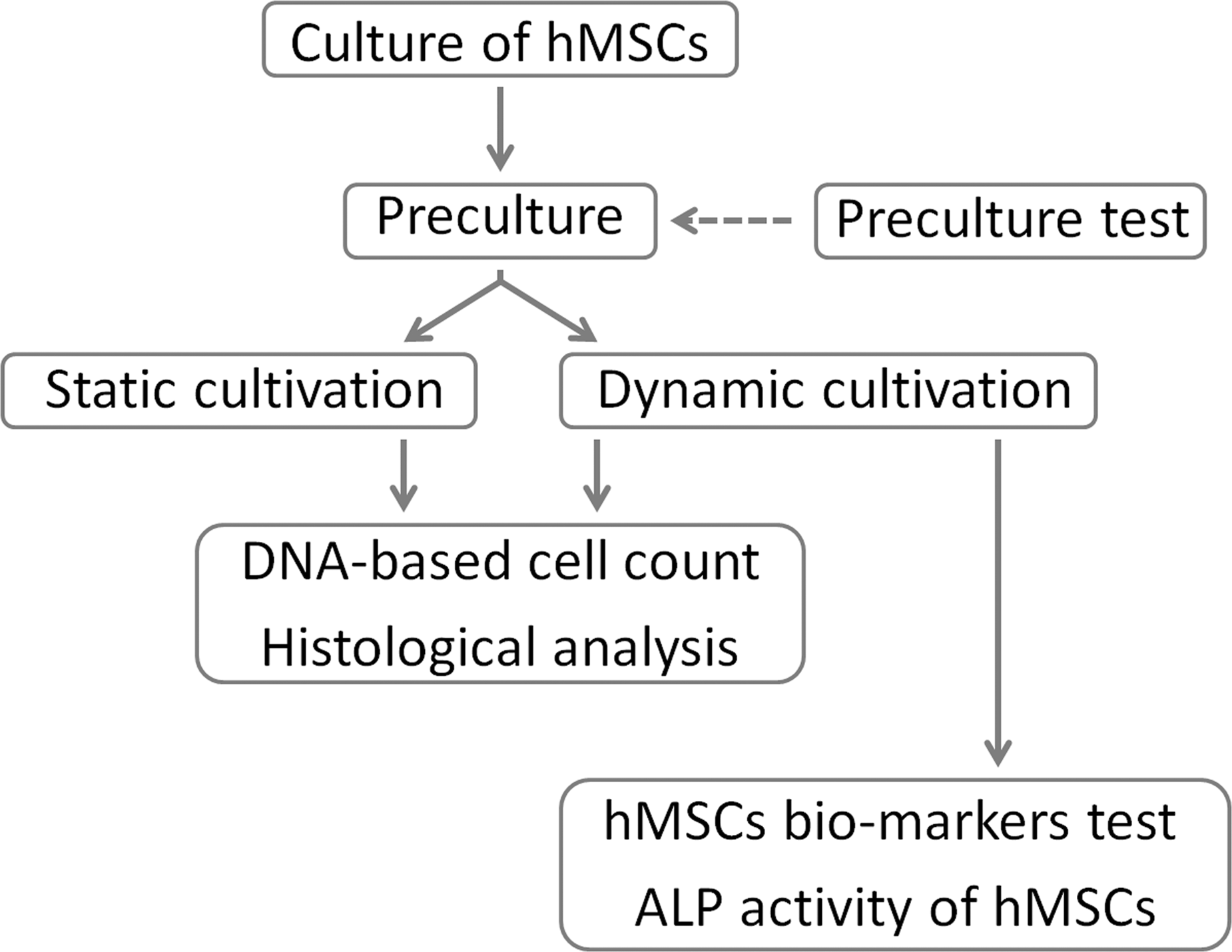
A flowchart of the present study.
Culture of human MSCs
MSCs derived from human bone marrow (PT-2501; Lonza Walkersville, Inc.) and donated by a 22-year-old man were passaged 5 times for use in this study. Dulbecco's modified Essential medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 100 unit/mL penicillin–streptomycin (Gibco) was used. Cell suspension containing 5×105 cells was seeded into 75-cm2 flasks, and 20 mL fresh culture medium added to each flask. Cultures were maintained at 37°C in a humidified atmosphere with 5% CO2. The culture medium was changed every 3 days. At 1-week cultivation, before reaching confluence, the cells were harvested by trypsin treatment and seeded onto type 1 collagen sheets (Gunze) (pore size, 70–110 μm; porosity, 80%–95%; diameter, 12 mm; thickness, 3 mm).
Preculture
It was necessary to optimize the incubation time to allow initial cell attachment to the collagen sheets. Therefore, a preculture assay was performed as described in a previous study. 12 Briefly, type 1 collagen sheets were placed in a 12-well plate, and cell suspension (80 μL) containing 1.15×105 cells was seeded onto them. The sheets were then incubated in a humidified atmosphere at 37°C with 5% CO2 for 3 or 6 h. Next, they were turned over, and a further 80 μL cell suspension was added before a further 3- or 6-h incubation. The sheets were then placed in 3 mL culture medium and cultured for 1 day. The number of cells dropping to the bottom of the well plate was examined.
Dynamic cultivation
Figure 2 shows the RFB (Able) and RFB cell culture system used. To form a scaffold, 3 precultured sheets were placed in the RFB in layers before incubation for 12 h (6 h+6 h). Temperature (37°C), pH (7.4), and dissolved oxygen (DO, 6.86 ppm) in the medium reservoir were controlled and monitored. The medium volume was maintained at 100 mL. After commencement of culture, the medium was changed every day from the third day onward. The medium flow rate was set at 3 mL/min. Culture was carried out for a total of 1 week.

Radial-flow bioreactor (RFB) system used in this study.
Static cultivation
The preculture protocol for static cultivation was the same as that for dynamic cultivation. An individual precultured sheet was placed in each well of a 12-well plate. The culture medium was maintained at 3 mL. Culture was carried out in a humidified atmosphere at 37°C and 5% CO2 for 1 week with no control of DO or pH values. The culture medium was changed daily from the third day of culture onward.
DNA-based cell count
At 1 week of culture, scaffolds with 3 cultured sheets from the RFB were selected for a DNA-based cell count. They were divided into upper, middle, and lower areas from top to bottom (Fig. 3A). Single collagen sheets were also selected from the static cultivation, because precultured collagen sheet was not laminated in static cultivation. This method of cell counting was selected based on an earlier study by this group. 12 Total DNA was quantified with the ND-1000 (NanoDrop Technologies). The cell number was then calculated using a working curve based on the cell number and total DNA.

The mean DNA-based cell count of the 3 areas under dynamic cultivation was compared with that under static cultivation.
Histological analysis
Histological analysis was carried out at 1 week of culture. The scaffolds in the RFB were divided into 9 areas horizontally and perpendicularly (Fig. 3A). With static cultivation, the sheets were divided into inside, middle, and outside areas (Fig. 3B). After embedding in paraffin, 4-μm-thick sections were prepared from both types of specimen and stained with hematoxylin–eosin. Finally, they were observed with an optical microscope.
The cells in the collagen sheet were visualized with antibodies of 4′,6-diamidino-2-phenylindole (DAPI) and phalloidin. Slides were incubated with 1% bovine serum albumin, DAPI (dilution 1:200), and phalloidin (1:100) for 30 min at room temperature. Next, the slides were washed in phosphate-buffered saline (5 min for 3 times). The sections were stained with DAPI and phalloidin, followed by observation under a laser-scanning microscope (LSM 5 DUO; Carl Zeiss). Three areas of 500×500 pixels each were selected randomly, and the cell nuclei counted using image analysis software (ImageJ 1.43u; NIH).
Real-time reverse transcriptase–polymerase chain reaction
To determine whether the characteristics of the hMSCs changed, real-time reverse transcriptase–polymerase chain reaction (RT-PCR) was carried out before seeding onto the collagen sheets and at after dynamic cultivation. After 1 week of culture, total RNA in the scaffold with real-time RT-PCR was stabilized using the RNAlater RNA Stabilization reagent (Qiagen). The hMSCs were then harvested, and total RNA was isolated using the RNeasy Plus kit (Qiagen) according to the manufacturer's protocol. Reverse transcription was carried out using QuantiTect Reverse Transcription (Qiagen). The gene GAPDH was used as an endogenous control for normalization of expression levels.
Real-time RT-PCRs were carried out using the ABI7500 (Applied Biosystems) with TaqMan gene expression assays (Applied Biosystems) Hs99999905_m1 (glyceraldehydes 3-phosphate dehydrogenase [GAPDH]), Hs00559595_m1 (CD29), Hs01075861_m1 (CD44), Hs00923996_m1 (CD105), Hs00233455_m1 (CD166), Hs02576480_m1 (CD34), and Hs00236304_m1 (CD45). Amplification was carried out as follows: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 95°C for 15 s and 60°C for 1 min. The 7500 fast system sequence detection software version 1.4. was used for data analysis.
Alkaline phosphatase activity in hMSCs after dynamic culture
To determine whether the hMSCs had retained the ability to differentiate after dynamic culture, the scaffolds were placed in a 12-well plate for 1 week, and 3 mL osteogenic induction medium was added to each well. The hMSC Differentiation BulletKit–Osteogenic (Lonza Walkersville, Inc.) was used as the osteogenic induction medium. Culture was maintained at 37°C in a humidified atmosphere with 5% CO2. The culture medium was changed every 3 days. At 2 weeks of cultivation, alkaline phosphatase (ALP) activity in the cells was determined. The scaffolds were rinsed with cold phosphate-buffered saline (PBS), cut into small fragments, and sonicated for 30 s after application of 200 μL Triton/PBS. The lysates obtained were centrifuged at 15,000 rpm for 15 min, and the supernatant was used as sample. ALP activity was assayed using LabAssay ALP (Wako) and determined by conversion of colorless p-nitrophenyl phosphate to colored p-nitrophenol. Phosphatase activity is proportional to the production of p-nitrophenol. Sample absorbance was measured in a 96-well plate at 405 nm. Amount of total protein in the sample was then examined with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.). Finally, ALP activity was expressed as units/μg protein.
Statistical analysis
Four samples were used for the DNA-based cell count. The number of nuclei in the scaffolds was determined by ImageJ. The DNA-based cell count was statistically analyzed using a one-way analysis of variance and the multiple-comparison Scheffe test (p=0.05). A two-way analysis of variance was carried out for the number of nuclei in the scaffolds, followed by a multiple comparison with the Scheffe test (p=0.05). Three samples were used to assess ALP activity in the hMSCs after dynamic culture. A t-test (p=0.05) was used in the analysis of the results for preculture and ALP activity.
Results
Preculture test
The number of cells dropping to the bottom of the well plate is shown in Figure 4. Fewer cells were observed on the bottom of the plate in the 12-h group than in the 6-h group (*p<0.05). Accordingly, precultured sheets incubated for 12 h (6 h+6 h) were used for both dynamic and static cultivation. The number of cells that dropped to the bottom of the well plate was considered to indicate the number of cells remaining in the collagen sheets; therefore, the smaller the number dropping, the larger the number remaining. The results showed that the seeding efficiency of this method was 88% in comparison to 2.3×105 cells at initial seeding.
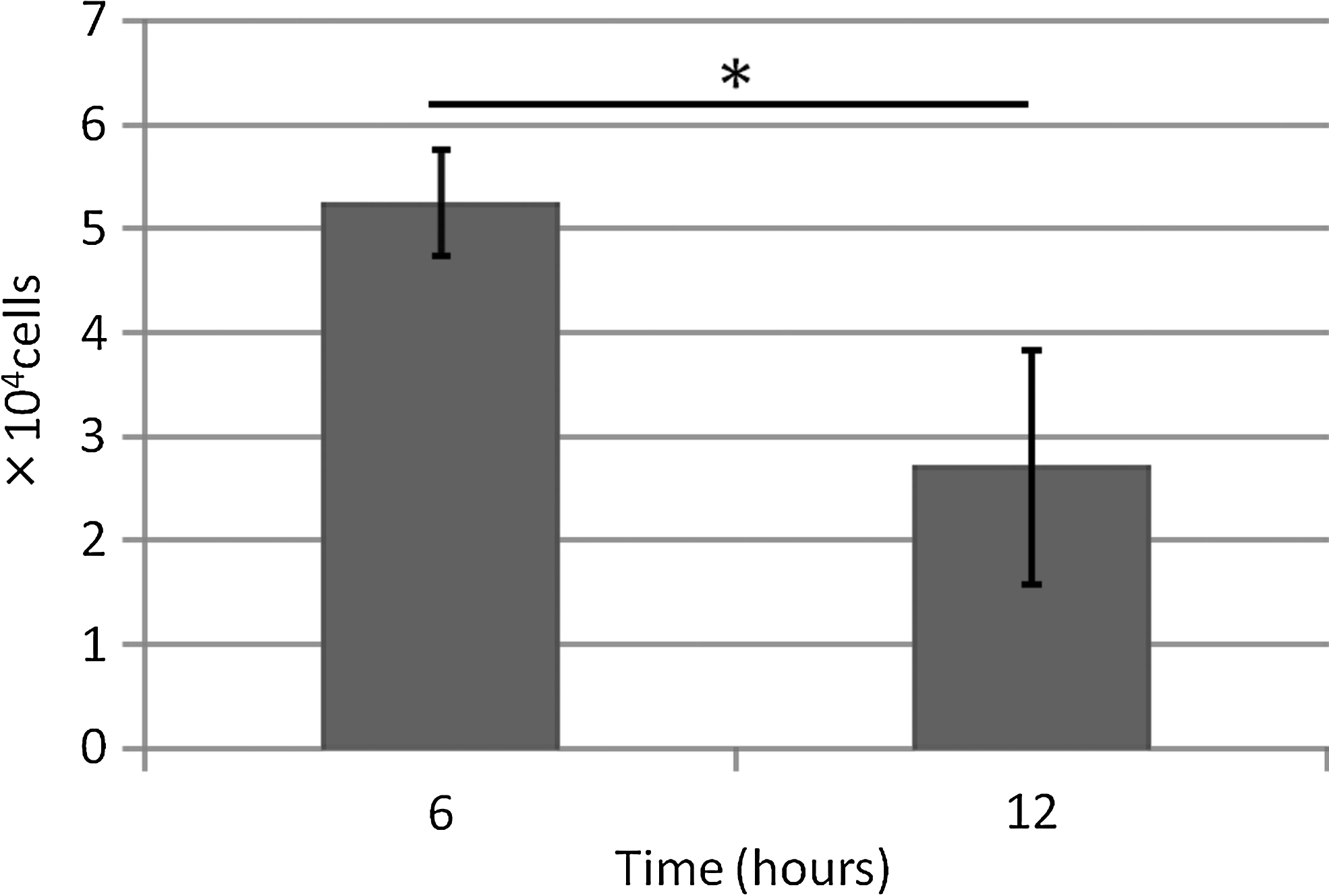
Number of cells that dropped to bottom of well plate. Significantly fewer cells were observed on bottom of plate in the 12-h group than in the 6-h group (*p<0.05). Data are expressed as mean±SD over 4 cultures.
DNA-based cell count
The DNA-based cell count is shown in Figure 5. With dynamic cultivation, the number of cells increased over that at initial seeding (2.3×105 cells) in all areas. No significant difference was observed in the number of cells between each area (Fig. 5A).
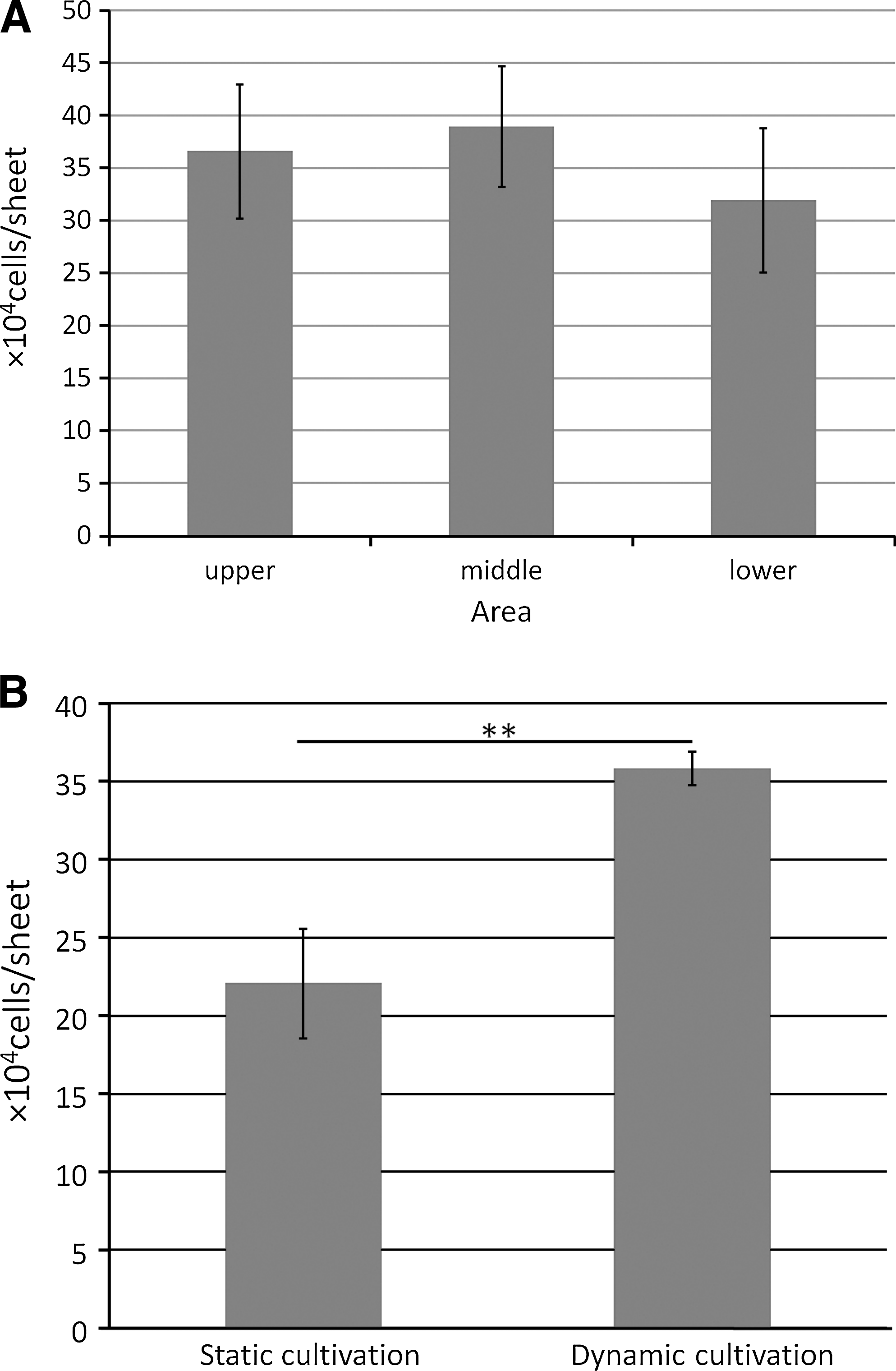
DNA-based cell count. Data are expressed as mean±SD over 4 cultures.
A significant increase was noted in number of cells under dynamic cultivation over that under static cultivation (Fig. 5B) (**p<0.01).
Histological analysis
Figure 6 shows optical micrographs of hematoxylin–eosin staining. A greater number of cells were observed in the collagen sheets under dynamic cultivation than under static cultivation. Visualization of the cells in the collagen sheet by staining with antibodies of DAPI and phalloidin is shown in Figure 7. These images also show a greater number of cells in the collagen sheets under dynamic cultivation in every area.
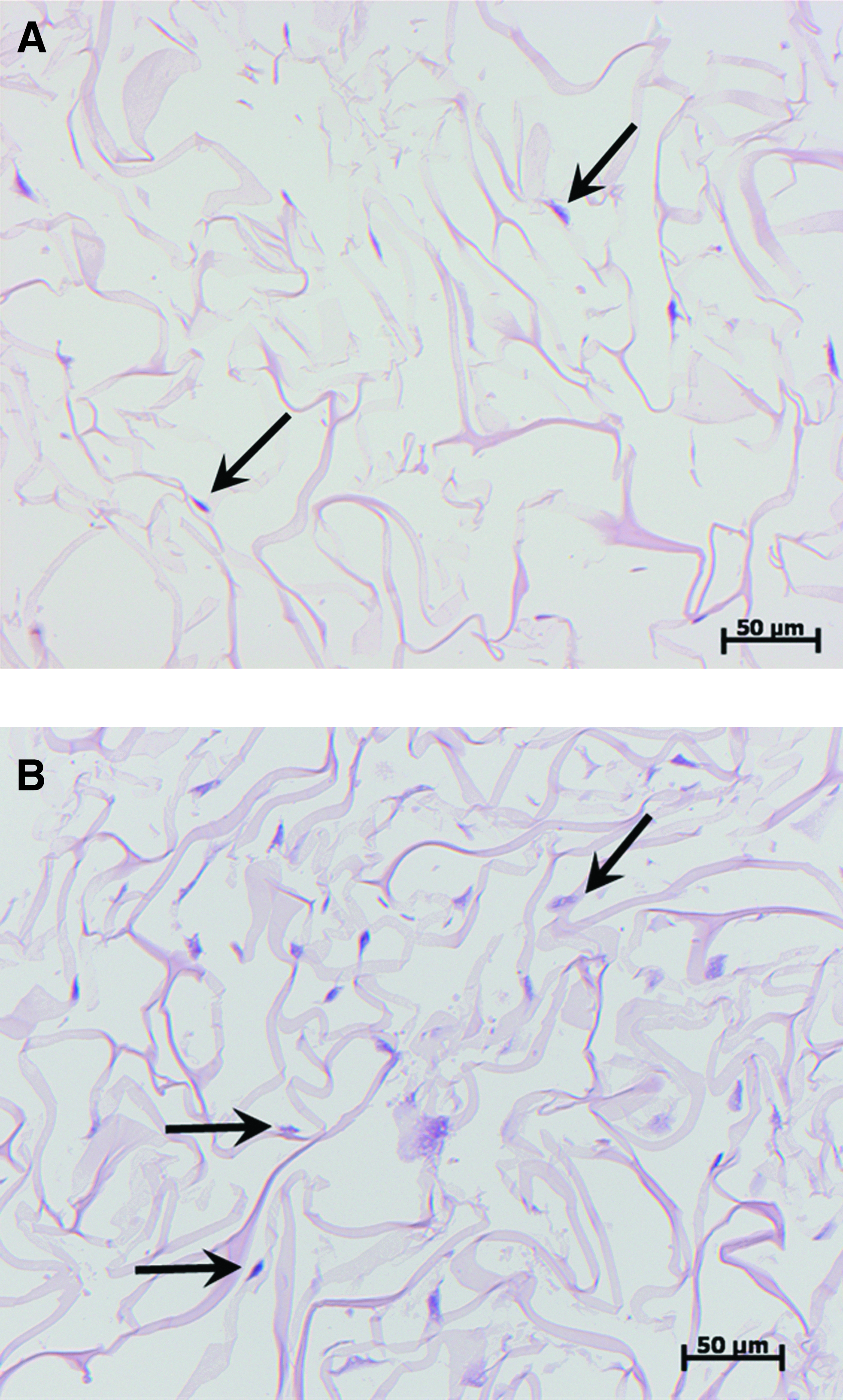
Typical optical micrographs of specimens stained with hematoxylin–eosin. (1 week)

Fluorescence micrographs of immunohistochemical staining with antibodies of 4′,6-diamidino-2-phenylindole (DAPI) (blue) and phalloidin (green). (1 week)
Figure 8 shows the number of cell nuclei under each type of cultivation as observed in fluorescence micrographs (Fig. 7). A significant difference was observed in the number of nuclei between each type of cultivation in each area (**p<0.01). On the other hand, no significant difference was observed in the number of nuclei between each area.
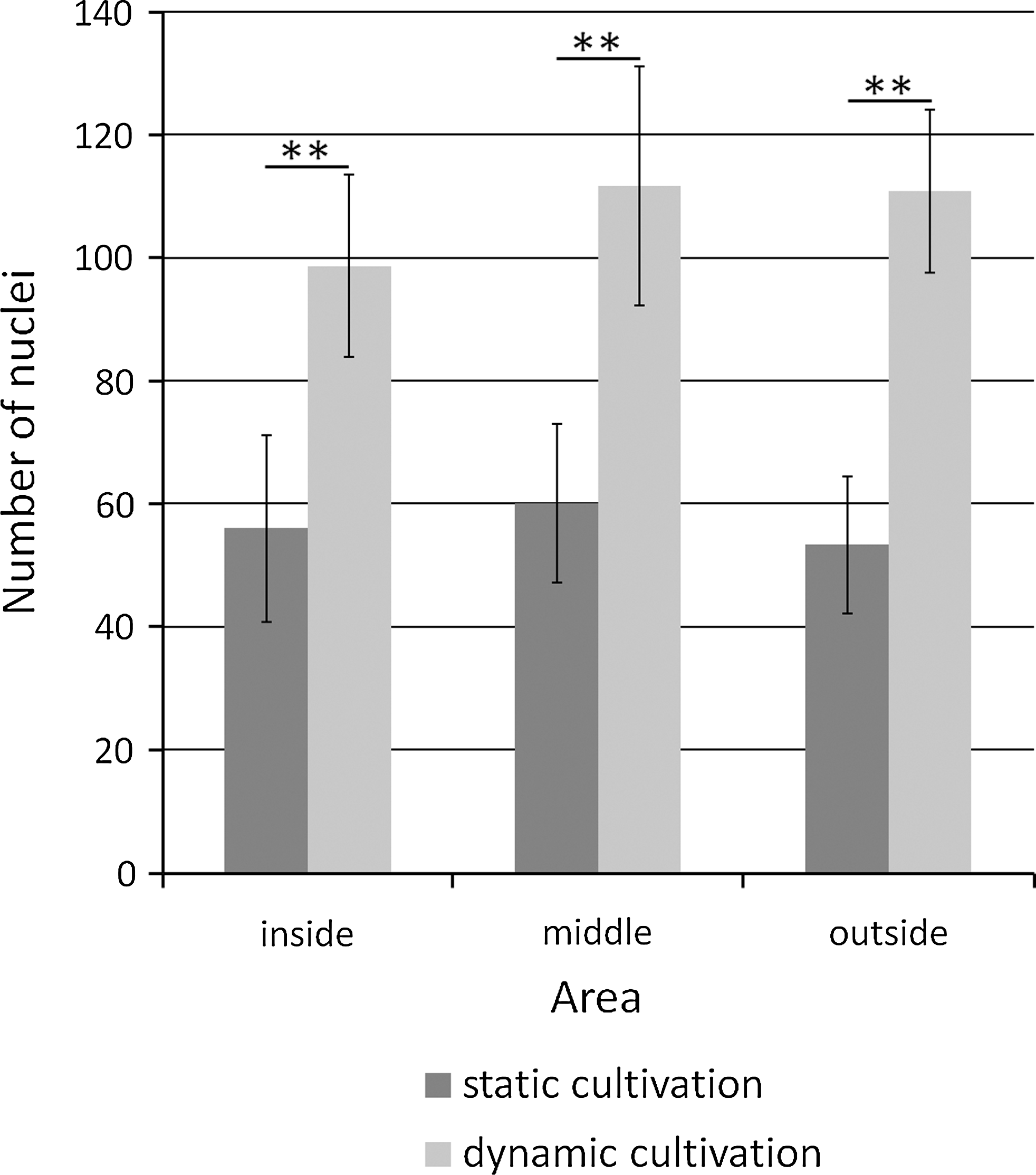
Number of nuclei in scaffolds under dynamic cultivation and static cultivation as determined by ImageJ. A significant difference in the number of nuclei was noted between dynamic and static cultivation (**p<0.01). No significant difference was noted in the number of nuclei among the inside, middle, and outside areas. Data are expressed as mean±SD over 4 cultures.
Real-time RT-PCR
Table 1 shows several hMSC markers from cells at before seeding onto collagen sheets and at after dynamic cultivation. No change was observed in these markers among these two time points.
No change was observed in these markers between these two time points.
ALP activity in hMSCs after dynamic culture
Figure 9 shows ALP activity after cultivation in an osteogenic induction medium. After 2 weeks, enzyme activity was significantly higher with cultivation in the osteogenic induction medium than in the DMEM.
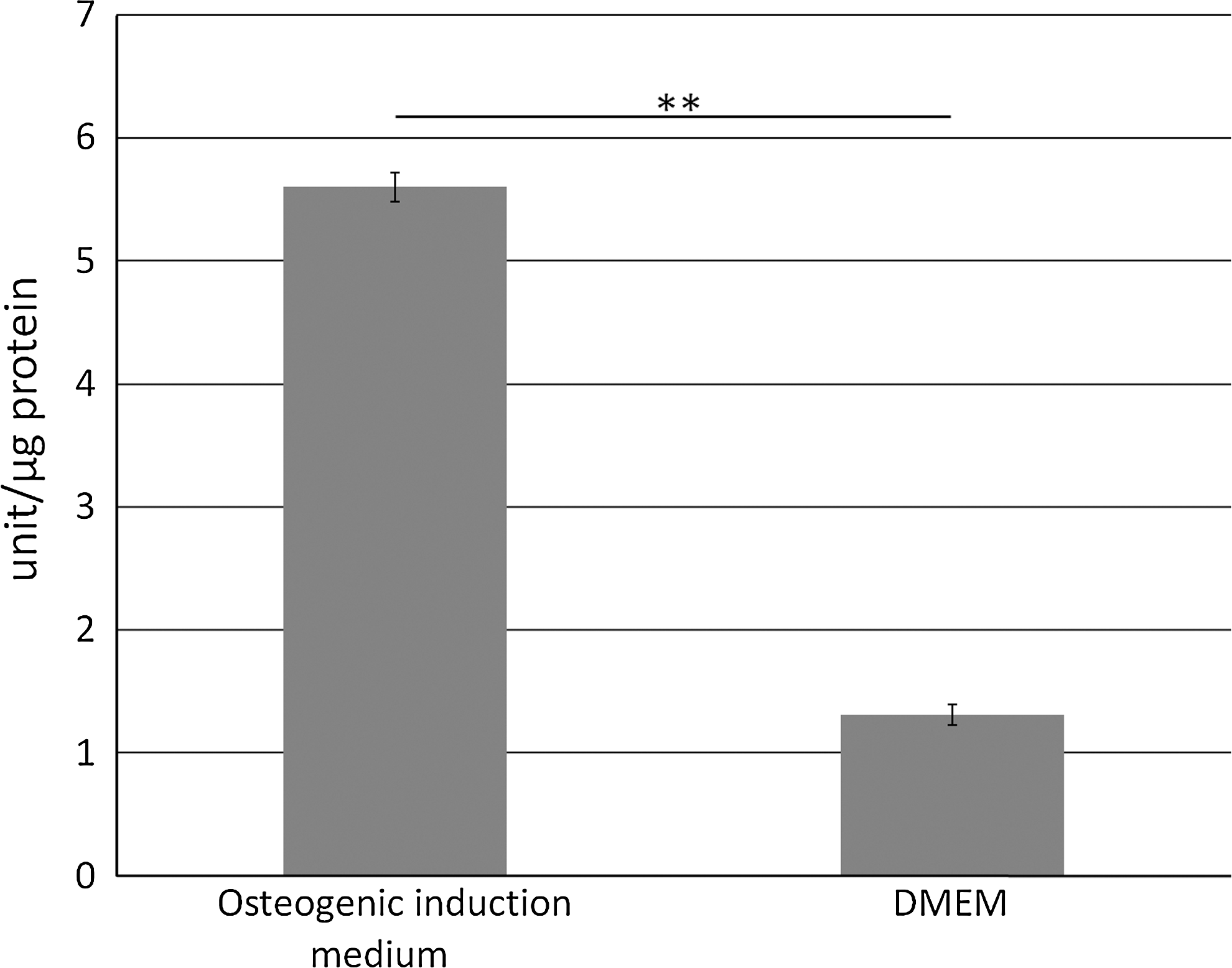
Alkaline phosphatase activity (units/μg protein) after cultivation in osteogenic induction medium. After 2 weeks, enzyme activity was significantly higher under cultivation in an osteogenic induction medium than under cultivation in the Dulbecco's modified Essential medium (DMEM) (**p<0.01).
Discussion
Bone tissue engineering using hMSCs is a promising new technique for bone regeneration. However, in vitro expansion of hMSCs on a 3D matrix is difficult. An RFB can maintain an even culture environment by providing a medium radially. Therefore, this study investigated whether an RFB enabled uniform expansion of hMSCs throughout a 3D scaffold with no change in cellular characteristics.
The cell number in the scaffolds was estimated by determining the quantity of DNA as a relative measure. In addition, histological analyses were performed using the cultured scaffolds. We set the culture period for 1 week based on an earlier study. 13 At 1 week of dynamic cultivation, a significant increase was observed in hMSCs. Distribution was also more uniform throughout the 3D scaffolds than that obtained with static cultivation. These results indicate the potential of the RFB as a system for 3D cultivation of hMSCs. This discrepancy in proliferation between the two types of cultivation may have been due to a more efficient delivery of nutrients and exchange of gas, along with the elimination of metabolic waste under dynamic cultivation.14,15 More oxygen is consumed in a 3D than in 2D culture. Moreover, the former provides a larger surface area, resulting in greater proliferation. The hypoxia and concomitant cell death usually associated with 3D culture are partially prevented by the use of a perfusion reactor. 16
Flow shear stress increases with an increase in the perfusion speed, which stimulates proliferation of cells and the formation of the extracellular matrix, including collagen, under dynamic cultivation. 17 However, the benefit of flow shear stress to the proliferation of hMSCs may depend on the flow rate and the type of bioreactor, cell, scaffold, or medium.18,19 The appropriate perfusion speed for the combination of materials used in this study remains to be determined. The speed was set as 3 mL/min, which was determined in reference to a previous report, 12 and an increase in cell proliferation was observed. The appropriate perfusion speed for hMSCs cultured in an RFB, therefore, remains to be determined.
The medium was replaced daily from the third day onward after initiation of culture based on the report of Arano et al. 12 Taking the number of cells seeded into account, 100 mL medium was used for dynamic cultivation, the minimum amount believed to be necessary based on preliminary experiments. No clear decrease was observed in the glucose concentration throughout the culture period. Furthermore, it would have been difficult to control the temperature, pH, or DO if the amount of the medium had been any smaller. The pH and DO were kept constant by continuous monitoring throughout dynamic cultivation to obtain stable results.
In general, antigens of human bone marrow MSCs are identified as positive for CD29, CD44, CD71, CD73, CD90, CD105, CD106, CD166, and Stro-1; and negative for CD11, CD14, CD18, CD31, CD34, CD40, CD45, CD56, CD80, and CD86. The hMSC markers were selected according to the manufacturer's (Lonza) information. Although not all the markers were investigated, those that were determined at two time points were at before seeding of cells onto the collagen sheet and at after dynamic cultivation. No change was detected in these markers between these two time points. At both time points, CD29, 44, 105, and 106 were positive, whereas CD34 and 45 were negative. These results were in accordance with those of a previous report, 20 indicating that no phenotypical change occurred in the cells between at before and after dynamic cultivation. Niehage et al. investigated an even larger number of surface molecules in hMSCs using a flow cytometer. 21 In this study, real-time RT-PCR was used, as it offered a simple means of determining cellular characteristics.
After culture in an osteogenic induction medium for 2 weeks, ALP activity was determined to evaluate the differentiation potential of the hMSCs under dynamic cultivation. ALP is a marker of early osteogenic differentiation and commitment of MSCs toward the osteoblastic phenotype. ALP activity was significantly higher with cultivation in an osteogenic induction medium than with cultivation in the DMEM. This result demonstrates that the hMSCs retained the potential for osteogenic differentiation after culture in the RFB. Further investigation of other osteogenic markers such as Runx2, type 1 collagen, OCN, and OPN is necessary, however, to confirm the differentiation potential of hMSCs. 21
For the in vitro clinical application of this technology, it is necessary to determine whether it is better to graft differentiated or nondifferentiated cells. As reported by Yoshinari et al., if hMSCs are cultured in an osteogenic induction medium using an RFB, the medium does not penetrate the scaffold equally due to calcification. 22 This can cause a number of problems such as cell death, nonhomogeneity in the scaffold, and sequestration after implantation. Therefore, in this study, we cultured hMSCs without an osteogenic induction medium in the RFB.
The present results showed that 3D culture in an RFB enabled uniform expansion of hMSCs with no change in cellular characteristics. The increase rate in hMSCs in this study however was lower than that of osteoblastic cells in our previous study. 12 This indicates the need to develop new approaches to improving the expansion ratio of hMSCs. The 3D culture of hMSCs in an RFB does however appear to offer potential in the clinical application of tissue engineering to the treatment of large bone defects.
Conclusions
The hMSCs expanded uniformly over a 3D scaffold under dynamic cultivation in an RFB with no change in cellular characteristics in comparison to static cultivation.
Footnotes
Acknowledgments
This research was supported by the Foundation of the Japan Medical Association, by the Oral Health Science Center Grant hrc7 from Tokyo Dental College, and by a “High-Tech Research Center” Project for Private Universities: matching fund subsidy from MEXT (Ministry of Education, Culture, Sports, Science and Technology) of Japan, 2006–2011. The authors would like to thank Dr. Yoshihiro Shibukawa, Dr. Hodaka Sasaki, Dr. Katsutoshi Kokubun, and Dr. Kaichi Matsuoka for their advice on this research, associate Professor Jeremy Williams for his assistance with the English of this article, and Mr. Tsuguyoshi Taira (Gunze Limited) for providing the collagen scaffolds.
Disclosure Statement
No competing financial interests exist.