
Abstract
Neural progenitor cells (NPCs) derived from human induced pluripotent stem cells (hiPSCs) can be differentiated to neural cells that model neurodegenerative diseases and be used in the screening of potential drugs to ameliorate the disease phenotype. Traditionally, NPCs are produced in 2D cultures, in low yields, using a laborious process that includes generation of embryonic bodies, plating, and colony selections. To simplify the process and generate large numbers of hiPSC-derived NPCs, we introduce a microcarrier (MC) system for the expansion of a hiPSC line and its subsequent differentiation to NPC, using iPS (IMR90) as a model cell line. In the expansion stage, a process of cell propagation in serum-free MC culture was developed first in static culture, which is then scaled up in stirred spinner flasks. A 7.7-fold expansion of iPS (IMR90) and cell yield of 1.3×106 cells/mL in 7 days of static MC culture were achieved. These cells maintained expression of OCT 3/4 and TRA-1–60 and possessed a normal karyotype over 10 passages. A higher cell yield of 6.1×106 cells/mL and 20-fold hiPSC expansion were attained using stirred spinner flasks (seeded from MC static cultures) and changing the medium-exchange regimen from once to twice a day. In the differentiation stage, NPCs were generated with 78%–85% efficiency from hiPSCs using a simple serum-free differentiation protocol. Finally, the integrated process of cell expansion and differentiation of hiPSCs into NPCs using an MC in spinner flasks yielded 333 NPCs per seeded hiPSC as compared to 53 in the classical 2D tissue culture protocol. Similar results were obtained with the HES-3 human embryonic stem cell line. These NPCs were further differentiated into βIII-tubulin+ neurons, GFAP+ astrocytes, and O4+ oligodendrocytes, showing that cells maintained their multilineage differentiation potential.
Introduction
It has been shown that neural progenitor cells (NPCs) and other neural cell types could be derived from iPSCs using various combinations of inhibitors of bone morphogenetic protein (BMP) and Activin/Nodal pathways, agonist of the Sonic Hedgehog pathway, or molecules such as retinoic acid (RA).10–13 One example of such a protocol is described by Nemati et al., 13 using a medium containing RA, noggin (a BMP pathway inhibitor), and fibroblast growth factor-2 (FGF-2) for 6 days followed by additional 6 days in the FGF-2 and RA medium, which resulted in the differentiation of iPSCs to NPCs. They were able to propagate these multipotent neural progenitors for a year. Kim et al. 12 showed that culturing ESCs and iPSCs in the presence of two small molecules, SB431542 and dorsomorphin, leads to neuroectodermal differentiation. They showed that the dual inhibition of the Activin/Nodal and BMP pathways could differentiate cell lines with diverse differentiation propensities.
iPSC-derived neural cells can potentially be used for cell replacement therapy, disease modeling, and drug screening and testing. Animal models are usually used to study neurological diseases. However, animals are imperfect models, because the animal disease phenotype may not correlate to those observed in humans due to the differences in genetics and anatomy. These neurological disease studies can also use hESCs carrying mutations of genes causing a few monogenic diseases, but not sporadic or complex neurological diseases.
Because of the relative ease of generating hiPSCs from the somatic cells of patients with neurodegenerative disease, hiPSCs were used in models of complex diseases such as amyotrophic lateral sclerosis, 14 familial dysautonomia, 15 spinal muscular atrophy (SMA) 16 and Rett syndrome. 17 These models allowed the identification of disease phenotypes that can be ameliorated by specific drugs. For example, Ebert et al. 16 observed that hiPSC-derived motor neurons from a child with SMA had low levels of survival motor neuron (SMN) protein aggregates called gems, and this was inversely correlated to the SMA disease severity. After treatment with valproic acid and tobramycin, SMN protein levels increased, and nuclear gems could be seen in these motor neurons. Their model can now complement animal models for screening potential drugs for SMA.
To have sufficient neural cells for drug testing, efficient cell expansion and neural differentiation protocols have to be created. A frequently used method of cell expansion for later differentiation to NPCs relies on 2D tissue culture (TC). In this method, the surface area for cell expansion is limited, and multistep medium feedings, plating, and cell selection are required for differentiation. Better scalability was achieved later when hESCs and hiPSCs were expanded on microcarriers (MCs)18–23 or as aggregates24,25 in a stirred spinner flask or bioreactors. Krawetz et al. 24 grew pluripotent H9 cells as aggregates derived from single-cell inoculum in the mTeSR™1 medium supplemented with Rho kinase inhibitor (Y-27632) and rapamycin. Their system achieved a cell yield of 4.5×105 cells/mL and a 25-fold cell expansion in a bioreactor. A similar expansion of pluripotent hiPSCs as aggregates (with evidence of some cystic structures) was reported by Olmer et al. 25 using mTeSR1 with Y-27632 in static plates; they reported a cell yield of 6.9×105 cells/mL and 23-fold expansion. The only work that describes expansion of iPSCs in MC culture is a review article written by Kehoe et al. 22 In it, the propagation of B12-3 hiPSCs on a Matrigel-coated MC in stirred spinners is reported with a cell yield of <4×105 cells/mL and sevenfold cell expansion. In summary, more work is needed to optimize and increase yields of hiPSC expansion in suspended cultures by one or more order of magnitude to produce enough neural cells for larger-scale drug testing. However, suspended cultures, even though they promise much higher scalability, have problems of their own, namely cell-damaging shear stress and absence of protocols of integration between expansion and differentiation stages leading to high yields of neural progenitors.
Shear stress on cells in stirred spinner flask and bioreactors reduces cell viability of mammalian cells. 26 Because shear stress can lead to loss of pluripotent cell markers, in addition to slower cell growth, it can affect the expansion of hESCs in stirred MC spinner cultures. 27 Yet, so far, its effect on iPSCs has not been investigated. Few studies have investigated large-scale expansion combined with complementary differentiation. Lock and Tzanakakis 21 have reported 10-fold hESC expansion and subsequent differentiation of these cells to definitive endoderm in MC spinner flask cultures. Steiner et al. 28 have shown that hESCs can be grown as aggregates in static plates achieving 2–5-fold expansion, and their differentiation could be controlled to become neurospheres. Serra et al. 29 have shown a 4.6-fold expansion and neural differentiation of the embryonal carcinoma stem cell line, Ntera-2/cl.D1, in a bioreactor.
In summary, although different groups have described some aspects of hiPSC and hESC expansion and/or neural differentiation in static or suspended cultures, there is still a major gap in developing efficient scalable dynamic systems in which hiPSCs can be expanded and directly differentiated to neural cells.
Our group has established a versatile MC platform for expansion of HES-318 and HES-227 in spinner flasks, achieving 17.5-fold and 12-fold expansion, respectively. This platform allowed the study of the metabolism of HES-3 cells grown on an MC in a serum or serum-free growth medium. 30 Furthermore, we have assessed that certain properties such as MC size, shape, coating, and positive charge are important for the long-term maintenance of hESCs on an MC. 31 We have also adapted this MC spinner flask system to generate cardiomyocytes from HES-3 and H1 cell lines. 32 These cardiomyocytes, when tested with the cardiotoxin Astemizole, prolonged their QT interval (the time between the start of the Q wave and the end of the T wave—a characteristic of depolarization and repolarization of the ventricles), demonstrating that they could be used for drug and toxicity screening.
Therefore, the aim of our study was to extend the use of this MC platform to generate hiPSC-derived NPCs that could be further differentiated to other neural cell types of potential utility for drug screening and disease modeling. We first modify the MC platform to expand iPSCs (IMR90) in suspension cultures. After the expansion, we direct the differentiation of these hiPSCs to NPCs on MCs in static cultures by simplifying a published protocol. 33 Lastly, we combine the expansion and neural differentiation on an MC in spinner flask suspension culture. The NPCs produced are further differentiated to cells expressing neuron, oligodendrocyte, and astrocyte markers.
Material and Methods
2D pluripotent stem cell maintenance, expansion, and neuroprogenitor differentiation
The induced pluripotent stem cell line, iPS (IMR90) (46, XX), was generously provided by James Thomson. 3 The hESC line HES-3 (46, XX) was obtained from ES Cell International. The cells were maintained on Matrigel- (Matrigel™ hESC-qualified matrix from BD Biosciences) coated TC plates (BD Falcon™) in an mTeSR1 (STEMCELL Technologies, Inc.) culture medium.
Matrigel was coated onto TC plates according to the recommendations. The medium was exchanged daily, and cells were passaged weekly at a split ratio of 1:15, using 1 mg/mL Dispase (STEMCELL Technologies, Inc.).
NPC differentiation from 2D cultures followed the schematic shown in Figure 1A. Following the protocol by Wu et al., 2012,
33
mTeSR1 was replaced a KnockOut™ (KO) medium, which consisted of 80% KnockOut DMEM, 20% KnockOut Serum Replacement, 1 mM
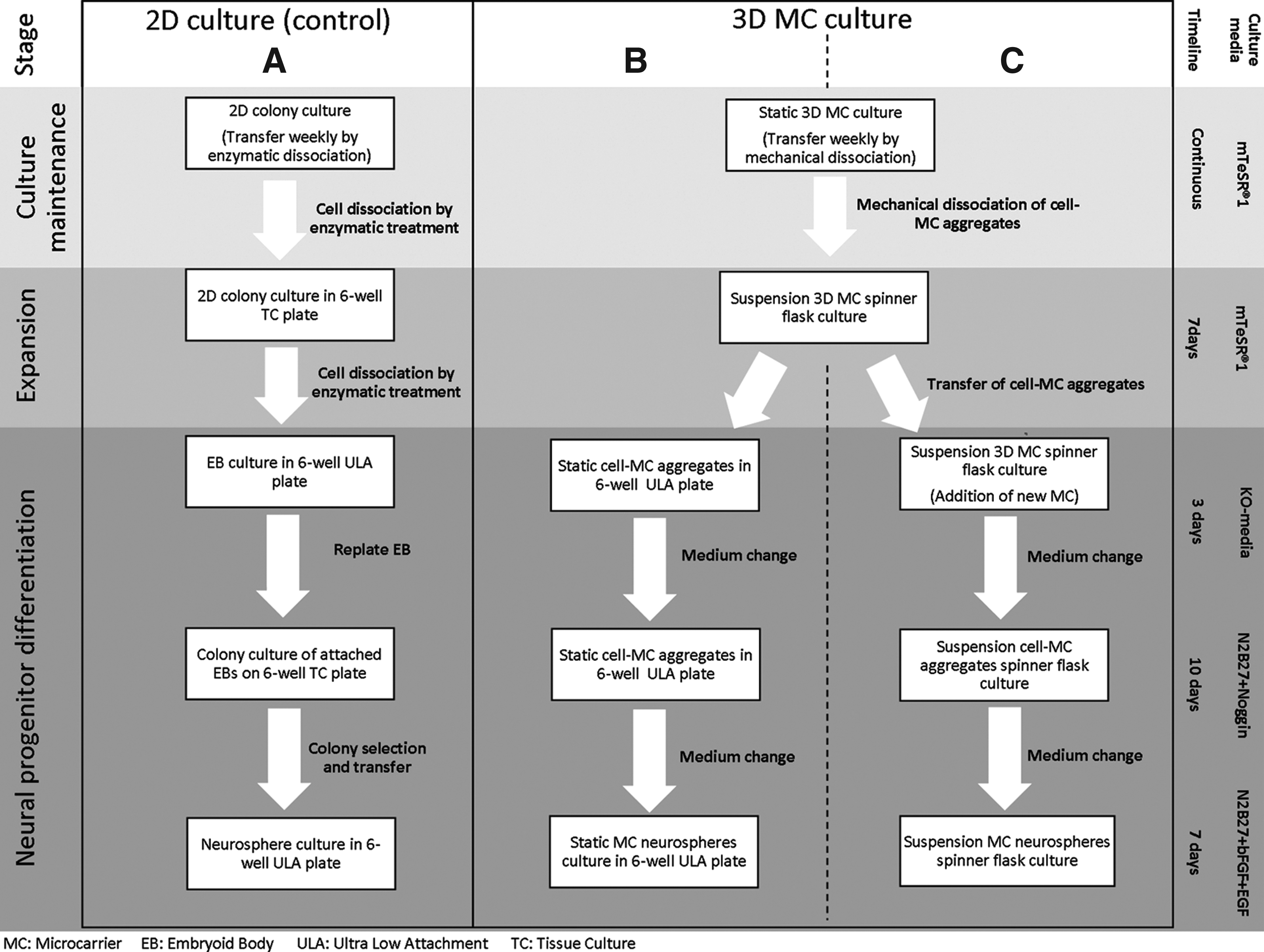
Schematic diagrams of the culture maintenance, expansion and differentiation process of induced pluripotent stem (iPS) (IMR90) cells to neural progenitor cells (NPCs) in 2D versus microcarrier (MC) systems.
3D MC static and suspension pluripotent stem cell cultures
DE-53 (Whatman) was selected as the MC for the culturing of iPS (IMR90) and HES-3 cells in suspension based on the method by Chen et al., 2011. 31 The preparation and sterilization of the MC are described previously in Chen et al., 2011. 31 Matrigel was diluted 72 times in ice-cold DMEM/F12 (Life technologies). Matrigel coating was prepared by adding 1 mL of the diluted Matrigel solution to 5 mg of the MC, followed by overnight agitation at 4°C.
Cultivation of iPS (IMR90) and HES-3 cells on MCs
To initiate MC culture, iPS (IMR90) or HES-3 cells on a TC plate were first enzymatically detached from TC plates using Dispase as described above. The cell concentration was adjusted to 8×105 viable cells/mL before seeding into a well of a six-well ULA plate (Corning Incorporated), containing 20 mg of Matrigel-coated DE-53 MCs and 4 mL of mTeSR1. The plate was shaken at 100 rpm for 1 h on a shaker in the incubator at 37°C/5% CO2; thereafter, the cells were grown in static conditions. Eighty percent of the medium was exchanged daily. For routine culture maintenance (see top section of Fig. 1B, C), 7-day-old cell–MC aggregate cultures were mechanically dissociated, and the viable cell concentration was determined using Nucleocounter NC-100™ (ChemoMetec A/S) before seeding into new six-well plates at the viable density of 1.6–1.8×105 cells/mL.
To expand iPS (IMR90) and HES-3 MC culture to a 100-mL spinner flask (BellCo Cat no. 1965-00100), a procedure similar to the one previously described by Chen et al., 2011, 31 was used. In brief, exponentially growing DE-53 MC static cultures were mechanically dissociated into small cell clumps and seeded at 2–3×105 cells/mL in the 100-mL spinner flask that contained 25 mL of mTeSR1 medium and 8 mg/mL of Matrigel-coated MC (Fig. 1B, C). The culture was kept static for 24 h in the incubator at 37°C/5% CO2, and thereafter another 25 mL mTeSR1 was added, and the culture was stirred at 25 rpm for 6 days. Eighty percent of the medium was exchanged once or twice daily. The viable cell concentration was determined using Nucleocounter NC-100. Cell–MC aggregate sizes were measured using the Olympus IX70 microscope at 4× magnification, and average sizes were determined using the associated software DP2-BSW version 2.2 (Olympus) following Lecina et al., 2010. 32 Metabolite analysis (glucose, lactate, ammonium, glutamine, and glutamate) and pH were determined by Bioprofile 100 plus (NOVA). Methods for calculating doubling time and specific metabolite consumption and waste product production rates were determined as described by Chen et al. 2010. 30
Spontaneous differentiation
iPS (IMR90) and HES-3 cells grown on the MC were spontaneously differentiated via in vitro EB formation according to Chin et al., 2007, 34 simply by changing the mTeSR1 to EB medium. 34 The medium was exchanged every other day for 21 days, and thereafter the differentiated cells were plated onto a 0.1% gelatin-coated TC plate and grown for 1–2 weeks and then immunostained for differentiation markers.
NPC differentiation in static and suspension MC cultures
iPS (IMR90) or HES-3 cells were propagated in an mTeSR1 medium in MC suspension spinner flask culture for 6–7 days to achieve pluripotent cell expansion; thereafter, NPC differentiation was carried out in static or suspension MC cultures as shown in either Figure 1B or C. For static MC cultures, cell–MC aggregates from spinner flask culture were simply transferred from the expansion medium to the differentiation medium in static six-well ULA plates following the same schedule and medium changes as described above for the 2D differentiation protocol, 33 but without doing any replating or manual colony selection steps (Fig. 1B). For differentiation in suspension spinner flask cultures (Fig. 1C), an additional 4 mg/mL of Matrigel-coated MC was added at the initiation of the differentiation process (day 0) to allow for further NPC expansion. Thereafter, these suspension MC aggregates were cultured following the same schedule and medium changes as described for the static MC cultures.
Neural differentiation
To demonstrate the multipotency of hESC- and iPSC-derived NPCs, 1–2×105 NPCs were seeded onto laminin-coated TC 12-well plates in an N2B27 medium without growth factors that differentiated them into neurons, astrocytes, and oligodendrocytes. For directed neuronal differentiation, the N2B27 medium was supplemented with 20 ng/mL brain-derived neurotrophic factor (BDNF; Peprotech, Inc.), 20 ng/mL glial cell line-derived neurotrophic factor (GDNF; Peprotech, Inc.), and 20 ng/mL nerve growth factor (NGF; Peprotech, Inc.). Neurons were allowed to form and mature for 3 weeks. The neurons' spontaneous postsynaptic currents and membrane potential were recorded by the standard patch-clamp protocol as described in a previous study by Wu et al., 2012. 33
RNA extraction and quantitative real time polymerase chain reaction
Cells from 2D TC or MC cultures were dissociated using TrypLE™ Express (Life technologies). MCs were removed from dissociated cells by passing the suspension through a 40-μm mesh (BD Falcon). Total RNA was then extracted and purified using RNeasy Mini Kit (QIAGEN) with on-column DNAse digestion (Qiagen). cDNA was synthesized from 500 ng of RNA per reaction following the protocol of SuperScript® II Reverse Transcriptase (Life technologies), and used in quantitative real-time polymerase chain reaction (qRT-PCR) reaction with the Power SYBR® Green PCR Master Mix (Life technologies). The list of QRT-PCR primers used can be found in previous studies by Chen et al., 2011, 31 and Wu et al., 2012. 33 Applied Biosystems 7500 Real-Time PCR System (Life technologies) was used for the subsequent reaction and analysis. Relative expression of various genes was determined by the ΔCt method with the expression of GADPH as the internal housekeeping reference in comparison to undifferentiated cells before differentiation.
Immunohistochemistry
Cells on the MC were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and stained either with pluripotent markers OCT3/4 (1:500; Santa Cruz) and TRA-1-60 (1:500, Millipore) or NPC markers Nestin (1:200; Covance) and paired box gene 6 (PAX6, 1:500; Developmental Studies Hybridoma Bank). Following that, secondary antibodies used were Alexa Fluor® 594 FluoroNanogold™ Fab’ fragment of goat anti-mouse IgG or Alexa Fluor 594 FluoroNanogold Fab’ fragment of goat anti-rabbit IgG (Life technologies).
Cells from EB- or NPC-differentiating cultures were plated on TC plates and fixed in 4% paraformaldehyde, and then they were washed with phosphate-buffered saline (PBS), permeabilized, and blocked with 0.1% Triton X-100/10% goat serum/PBS. The respective primary antibody was added and left overnight at 4°C. The following primary antibodies were used: mouse monoclonal antibody against α-fetoprotein (AFP, 1:250; Sigma-Aldrich, Inc.), mouse monoclonal antibody against α-smooth muscle actin (α-SMA, 1:400; Sigma-Aldrich, Inc.), mouse monoclonal antibody against PAX6 (1:500; Developmental Studies Hybridoma Bank), mouse monoclonal antibody against βIII-tubulin (1:500; Chemicon), mouse monoclonal antibody against Oligodendrocyte marker O4 (O4, 1:100; Chemicon), rabbit monoclonal antibody against glial fibrillary acidic protein (GFAP, 1:500; Dako), and rabbit polyclonal antibodies against Nestin (1:250; Covance). The cells were washed with 1% bovine serum albumin (BSA) in PBS, and secondary antibodies were added for 1 h in the dark at room temperature. The following secondary antibodies were used: Alexa Fluor 488 FluoroNanogold Fab’ fragment of goat anti-mouse IgG or Alexa Fluor 594 FluoroNanogold Fab’ fragment of goat anti-rabbit IgG (Life Technologies). Cells were washed (1% BSA solution) and imaged using an inverted Olympus IX70 fluorescent microscope (Olympus).
Flow cytometry
The expression levels of extracellular markers TRA-1-60 and mAb84 35 and transcription factor OCT3/4 in iPS (IMR90) were measured by flow cytometry as described previously by Oh et al., 2009. 18 Similarly, expression levels of the polysialic acid form of neural cell adhesion molecule (PSA-NCAM)-positive NPCs were quantified using flow cytometry with PSA-NCAM APC-conjugated monoclonal antibody (1:10; Miltenyi Biotec GmbH).33,36 Gating was set at the point where positive and negative cell populations intercept. Positive cell population was then determined by subtracting the negative population within the gate. 18
Karyotype analysis
iPS (IMR90) cells grown on MCs for more than 10 passages were sent to Cytogenetics Laboratories at the Department of Obstetrics and Gynaecology, Kandang Kerbau Women's and Children's Hospital, Singapore, for karyotype analysis, as described previously. 18 Karyotype analysis was performed with 20 cells.
Statistical analysis
All experiments were performed at least in triplicates. Data values are reported as a mean and standard deviation. These analyses and graphs were done in Microsoft® Excel® 2008. Student's t-test analysis of variance was applied to compare between groups, and p<0.05 was considered as significant.
Results
Serial expansion of iPS (IMR90) in static MC culture
iPS (IMR90) cells derived from human fetal fibroblasts by Yu et al., 2007, 3 can be propagated in 2D Matrigel-coated culture in an mTeSR1 serum-free defined medium. These hiPSCs maintained pluripotency during extensive passaging. 3 Cell densities of 1.8×106 cells/mL and 11.3-fold expansion are achieved. Obtaining larger amounts of iPSCs in 2D cultures is hampered due to the restricted surface area for cell growth. One of the ways of overcoming this limitation is a 3D MC system in which cells are grown on the surface of small particles in agitated bioreactors under dynamic conditions rather than on 2D TC surfaces.
Since the DE-53 cellulose MC platform had been shown to support cell growth in a conditioned or defined medium (mTeSR1, StemPRO® hESC SFM) of HES-318,30 and HES-2 27 cells, we used it to test the ability of the MC to support expansion of pluripotent iPS (IMR90). The cells harvested from 2D TC plates were transferred to six-well ULA plates containing Matrigel-coated DE-53 MCs. These cells were propagated for 7 days in an mTeSR1 medium and passaged 10 times by mechanical dissociation of cell–MC aggregates, at a seeding density of 1.6×105 cells/mL. Over the first 10 passages, the cell density achieved on day 7 ranged 1–1.6×106 cells/mL with an average of 1.3±0.03×106 cells/mL and 7.7±0.2-fold expansion (Fig. 2A). Flow cytometry analysis showed that the pluripotent markers OCT3/4 and TRA-1-60 were maintained during the 10 passages at 69.9%±2.9% and 84.0%±7.2%, respectively. These expression levels are similar to the controls obtained with iPS (IMR90) grown on 2D TC plates (OCT3/4 72.5%±13.9%, TRA-1-60: 88.1%±8.4%), indicating that they retained pluripotency (Fig. 2A). The pluripotent state of these cells was further confirmed by immunohistochemistry of the cell–MC aggregates, which showed an evenly distributed positive staining with OCT3/4 and TRA-1-60 markers of the cells surrounding the MC (Fig. 2B).
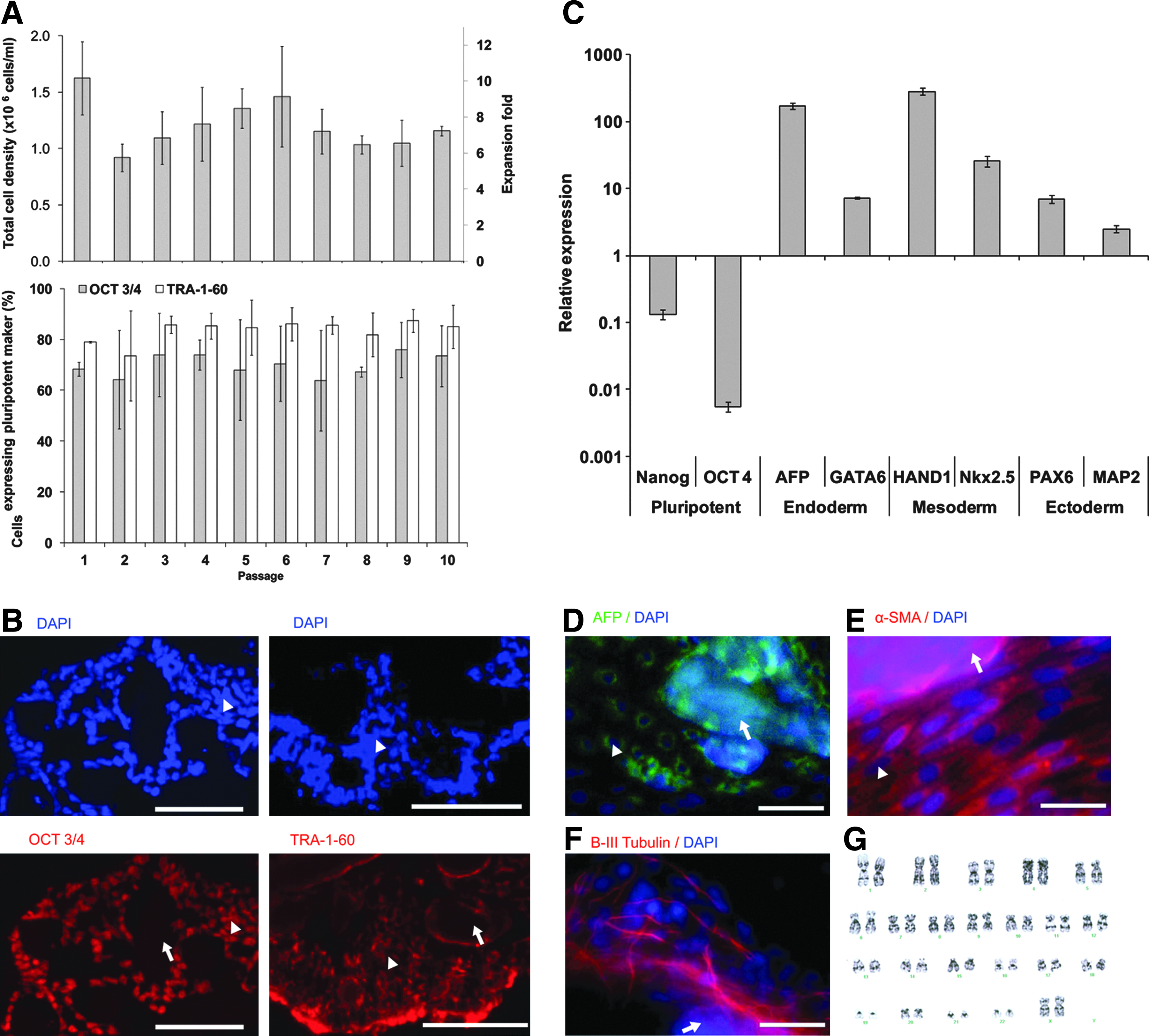
Long-term expansion of pluripotent iPS (IMR90) cells grown on human embryonic stem cell (hESC)-qualified Matrigel™-coated DE-53 MC.
To explore whether iPS (IMR90) cells grown on the MC kept their ability to differentiate into derivatives representing the three germ layers, cells from MC cultures (passage 10) were spontaneously differentiated into EBs. qRT-PCR analysis of mRNA transcripts showed a downregulation of expression of the representative pluripotency markers Nanog and OCT3/4 and upregulation of representative endoderm markers AFP and GATA6, mesoderm markers HAND1 and Nkx2.5, and ectoderm markers PAX6 and MAP2 (Fig. 2C). EB-derived cells were also analyzed with immunocytochemistry and were found to stain positive for AFP (endoderm), α-SMA (mesoderm), and β-III tubulin (ectoderm) (Fig. 2D–F). Also, karyotype analysis of iPS (IMR90) cells propagated in MC culture for 14 passages showed that cells maintained a normal karyotype (Fig. 2G).
Scaling up of iPS (IMR90) MC cultures in stirred spinner flask
MC cultures in static conditions are limited in growth and scaling-up abilities due to their non homogeneous nature. Cells within the large cell–MC aggregates (Fig. 3B) generated in static conditions may have limited access to nutrient supply. This limitation can be overcome in stirred homogeneous MC cultures. To this end, iPS (IMR90) cells from static MC cultures were seeded at a density of 3×105 cells/mL into a 50-mL stirred spinner flask containing 4 mg/mL of DE-53 MC. As controls, cells were also seeded into 2D TC and static DE-53 MC cultures. Cell growth kinetics, culture morphology, and levels of pluripotency marker expression were monitored in all three conditions (Fig. 3D, E).
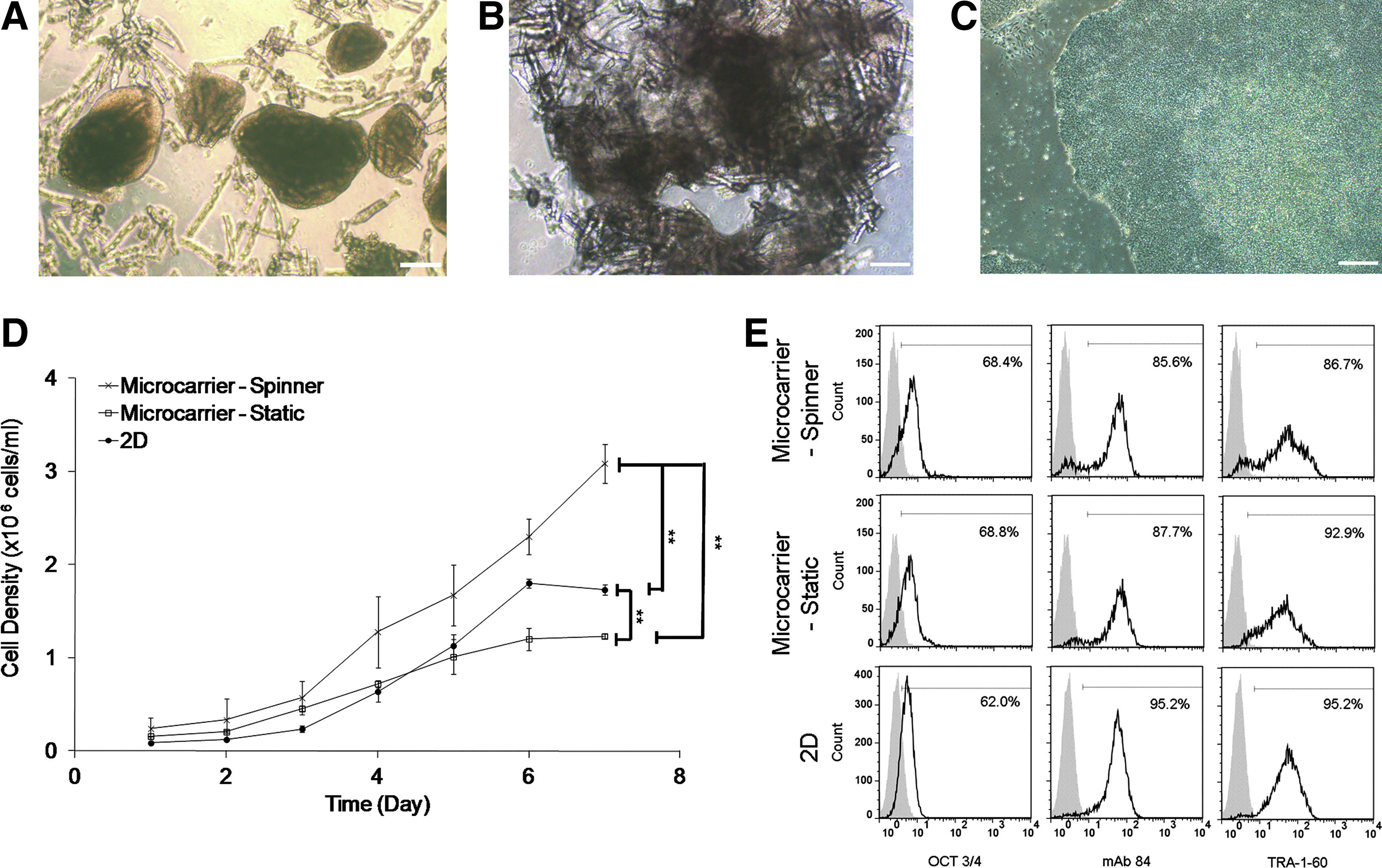
Expansion of iPS (IMR90) in 2D and 3D static and spinner MC cultures. Representative images of iPS (IMR90) cells
Exponential cell growth started between day 2 and 3 in all conditions, and the maximum cell density was reached between day 6 and 7 (Fig. 3D). Maximum viable cell density achieved in the spinner flask (3.1±0.2×106 cell/mL, viability of 90.6%±6.1%) was significantly higher than in static six-well plates (1.23±0.03×106 cells/mL, viability of 99.2%±2.1%) and on 2D TC plates (1.80±0.05×106 cells/mL, viability of 99.4%±3.3%). The doubling time of iPS (IMR90) cells during the exponential phase in stirred MC cultures (35.8±7 h) was shorter than in static MC culture (41.7±6.8 h), but similar to the TC control condition (33.2±5.8 h) (Table 1).
PSA-NCAM, polysialic acid form of neural cell adhesion molecule; NPCs, neural progenitor cells; hESC, human embryonic stem cell; hiPSCs, human induced pluripotent stem cells.
On 2D TC cultures, dense cell colonies were observed (Fig. 3C). In the spinner flask culture, iPS (IMR90) cell–MC aggregates measured on day 7 had an average height and width of 396±125 μm and 296±94 μm, respectively (Fig. 3A). In comparison, in static MC cultures, cell–MC aggregates were much larger and more heterogeneous (Fig. 3B). The smaller aggregate sizes and more homogeneous culture may explain the improvement in cell yields and doubling times in spinner versus static MC cultures (Fig. 3D).
Levels of expression of the pluripotent markers OCT3/4, mAb84, and TRA-1-60 were similar in all conditions (Fig. 3E). However, the small secondary peaks seen in the histograms of mAb84 and TRA-1-60 in MC spinner cultures may indicate the presence of a small population of differentiating cells, which may be due to the shear stress.
Effect of feeding regime on cell yield and pluripotency of iPS (IMR90) in spinner flask MC cultures
Metabolic analysis of iPS (IMR90) DE-53 spinner flask MC cultures showed that during the last days of propagation (days 6–7), lactate was accumulated at high levels of about 2 mg/mL leading to a low pH of about 6.5 (data not shown). A previous study by Chen et al., 2010, 30 suggested that in hESC cultures, lactate concentration above 1 g/L could significantly reduce cell growth and induce differentiation. To test if we could achieve higher cell yields in hiPSC MC cultures, the growth medium exchange rate was increased from once to twice a day, which resulted in an increase in pH and lower lactate levels. As shown in Figure 4A, in both spinner flasks (once- or twice-daily feeding), the exponential cell growth started on day 3 reaching on day 7, a viable cell density of 3.1±0.2×106 cells/mL (viability of 90.6%±6.1%), and 6.1±0.57×106 cells/mL (viability of 96.2%±3.4%), respectively. The doubling time of cells in the twice-daily-fed spinner (31.8±4.1 h) was shorter than the once-daily-fed culture (35.8±7 h) (Table 1). Moreover, beyond the twofold increase in the cell density, the change in feeding regime from once to twice daily also increased the expression of pluripotency markers OCT3/4, mAb84, and TRA-1-60, which were equivalent to those grown on 2D TC plates (Fig. 4D). The secondary peaks seen in the histograms of the once-daily medium exchange were not seen in the twice-daily medium exchange conditions, indicating that the higher pH and lower lactate concentration in twice-daily-fed cultures probably led to the higher pluripotent state.

iPS (IMR90) cell growth in once- versus twice-daily medium-exchange stirred spinner flask cultures.
Microscopic observation of the culture showed more MC–cell aggregates in the twice-daily-fed cultures (Fig. 4B) compared to the once-fed cultures (Fig. 3A). Larger cell–MC aggregates were obtained, and the average length and width was 596±210 μm and 474±185 μm, respectively, for the twice-fed cultures as compared with 396±125 μm and 296±94 μm for the once-daily-fed condition. Moreover, fewer empty carriers were seen in these twice-daily-fed cultures. Immunohistological sections of iPS (IMR90) cell aggregates showed positive staining for the OCT3/4 marker (Fig. 4C).
These findings indicate that at a high cell density, growth may be limited as a result of accumulation of toxic products (maybe lactate), environmental effects (maybe low pH), or unknown nutrient or growth factor limitations. These limitations, which may also affect levels of pluripotency, can be overcome by manipulating feeding regimes (e.g., control of low glucose levels). Further studies are needed to clarify the above issues.
Differentiation of iPS (IMR90) cells to NPCs in static MC cultures
The current 2D culture platform for NPC differentiation has limited scalability 33 (Fig. 1A). The NPC differentiation procedure was based on generating EBs in a KO medium followed by replating the EBs to the TC plate in a Noggin-supplemented N2B27 medium and finally generating neurospheres by manual selection of rosettes and continuation of the differentiation process in an FGF-2- and EGF-supplemented N2B27 medium. 33 This process usually lasts 20 days and generates about 85% PSA-NCAM+ NPCs and a 4.7-fold NPC expansion (Table 1). The presence of NPCs was confirmed also by immunostaining of PAX6 and Nestin. 33
After the development of an efficient MC expansion platform, we continued to direct the differentiation of hiPSCs into NPCs, first in static MC conditions (Fig. 1B) using the same medium-feeding regime. Cells expanded in twice-fed MC spinner culture conditions were transferred directly into static six-well plates for differentiation (Fig. 1B). As a control, 2D TC expansion and differentiation process were carried out also in six-well plates (Fig. 1A). On day 20, at the end of the differentiation process, rounded neurospheres were formed in both conditions (Fig. 5A–D); the average length and width of MC-derived neurospheres (L: 835±212 μm×W: 750±102 μm) were significantly larger than those derived from 2D controls (L: 485±231 μm×W: 420±188 μm). It is important to note that MCs were embedded within all the neurosphere cell–MC clumps (Fig. 5A, C)

Differentiation of iPS (IMR90) to NPCs in static MC versus 2D culture conditions. Representative images of neurospheres on day 20 generated from
To determine the yields of NPC generation in both conditions, the percentage of PSA-NCAM+ NPCs was determined by flow cytometry, and the cell density was measured by the nucleus count method. PSA-NCAM is a known surface marker of NPC. 36 Volumetric yields of NPCs were calculated by multiplying the percentage of expression of PSA-NCAM+ cells with the final cell concentration. Both processes had similar PSA-NCAM+ expression levels and final cell yields (Fig. 5J). The volumetric NPC yield was 2.2±0.47×106 NPCs/mL in MC culture, as compared with 2.3±0.14×106 NPCs/mL in the TC conditions. The single cells obtained from the trypsinized neurospheres from both systems exhibited more than 95% viability as tested by the trypan blue exclusion method (data not shown).
Taken together, the overall yield of NPCs (Fig. 5K) at the end of the differentiation process per cell seeded at the beginning of the expansion phase is about twofold higher in the static MC culture condition (Fig. 1B) than in the 2D TC condition (Fig. 1A and Table 1).
To determine if the MC culture-derived differentiation process is able to generate NPCs of the same quality as 2D TC-derived NPCs, 20-day-old NPC neurospheres from both differentiation systems were plated onto laminin-coated plates and stained for the neural progenitor markers, PAX6 and Nestin. Both the MC- and TC-derived cells stained positive for PAX6 (Fig. 5E, F) and Nestin (Fig. 5G, H). Furthermore, qRT-PCR analysis of mRNA transcripts for the neural progenitor markers, PAX6, SOX1, Nestin, and MSI also showed a similar upregulation of these transcripts in cells from both processes relative to undifferentiated cells (Fig. 5I). The qRT-PCR analysis also shows upregulation of transcripts of representative endoderm and mesoderm markers, AFP and HAND1, respectively, indicating that under both MC and TC conditions, some differentiation into non-neural cell types might also have taken place.
Kinetics of iPS (IMR90) differentiation to NPCs in stirred MC cultures
After demonstrating the feasibility of generating PSA-NCAM+ NPCs in static MC cultures, the next step was to accomplish a continuous expansion and differentiation process in the same stirred spinner flask (Fig. 1C). To this end, iPS (IMR90) cells were first expanded in MC spinner flasks with twice-daily medium exchange regime for 7 days and then reseeded at 2×105 cells/mL to MCs in spinner flasks for further NPC differentiation (Fig. 1C). At the beginning of the differentiation process, 2 mg/mL of Matrigel-coated MCs was added to support further cell expansion. Cell growth kinetics and PSA-NCAM+ expression were monitored between days 7 and 20 of the differentiation process. The maximal cell density of 10.6×106 cells/mL was obtained on day 18 (Fig. 6A), followed by a decline in the cell concentration on day 20. Flow cytometry analysis for PSA-NCAM expression showed that the highest percentage of PSA-NCAM+ cells (78%) was achieved also on day 18 (Fig. 6B). Thus, on day 18, the volumetric yield of PSA-NCAM+ NPCs was 8.3×106 cells/mL (Fig. 6C). The overall yield of NPCs obtained after the differentiation process per hiPSC seeded at the start of the expansion phase was 333 NPCs/hiPSC (Fig. 6D), which is about 3 times higher than the static MC cultures (93 NPCs/hiPSC) (Fig. 1B and Table 1) and about 6 times higher than the 2D TC (53 NPCs/hiPSC) (Fig. 1A and Table 1).
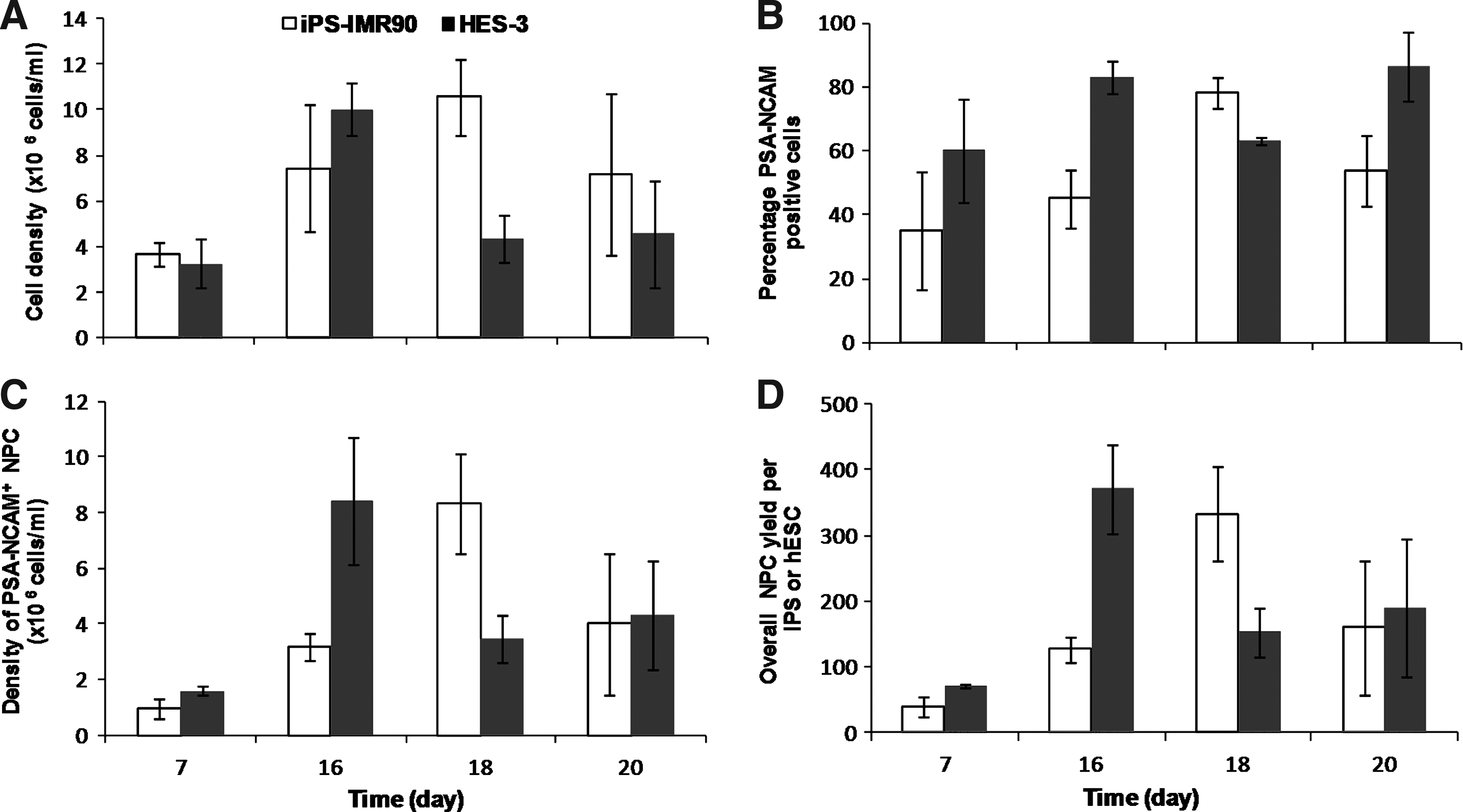
Kinetics of iPS (IMR90) hiPSC and HES-3 hESC differentiation to NPCs in spinner flask MC cultures.
Expansion and differentiation of hESCs to NPCs in stirred MC cultures
To validate the universality of the MC expansion and differentiation-based protocol, we tested if it could be applied to the HES-3 hESC line. The expansion of HES-3 cells in Matrigel-coated DE-53 MCs was described previously by our group.18,30,31 HES-3 cells were successfully propagated in static cultures for many passages in a variety of serum-containing and serum-free media without losing pluripotency and achieving cell yields ranging 1.5–2×106 cells/mL. When grown in an mTeSR1 medium, static MC culture achieved a cell density of 1.54±0.09×106 cells/mL, 15.4±0.9-fold cell expansion, and doubling time of 35.8±2 h. 30 Propagation in spinner flask cultures with a conditioned medium led to a high density of 3.5×106 cells/mL; however, the cells showed a tendency to differentiate presumably as a result of the shear stress.18,27
HES-3 expansion in MC spinner flasks with a twice-daily-fed mTeSR1 medium reached a cell density of 4.3±0.01×106 cells/mL, and a 21.3±0.3-fold increase in cell yield was achieved on day 7, which is approximately double the yield of single-feed MC spinner and 2D TC cultures (Table 1). While HES-3 cells grown in a conditioned medium had the tendency to undergo spontaneous differentiation under agitation condition, 27 pluripotency was maintained (84.6%±1.65% OCT3/4 and 96.8%±2.3% mAb84) in the mTeSR1 medium.
HES-3 MC culture was further differentiated to NPCs using the protocol described above (Fig. 1C). Cell growth and PSA-NCAM+ NPC generation kinetics were investigated (Fig. 6). Similar to the results obtained with hiPSCs, maximal yields were obtained on day 16 of growth followed by a drop in cell concentration thereafter (Fig. 6A). On day 16 of the differentiation process, a cell concentration of 8.4×106 cells/mL PSA-NCAM+ NPCs and a further 17.7-fold NPC expansion were achieved (Table 1). The overall yield of the expansion and differentiation process was 371 PSA-NCAM+ NPCs per seeded HES-3 cells, which is 11.6-fold higher than the 2D differentiation process (32 NPC/hESC). This improved yield is similar to the one obtained with hiPSCs (333 NPC/hiPSC), which validates the robustness of our novel expansion and differentiation protocol for NPCs.
Characterization of iPS- (IMR90) and HES-3-derived NPCs and their differentiation to the three neural lineages
hiPSC- and hESC-derived neurospheres obtained from the integrated expansion and differentiation spinner flask process (Fig. 1C) were further evaluated for NPC marker expression and their ability to generate cells expressing neuronal, astrocyte, and oligodendrocyte markers—the three neural lineages.
iPS (IMR90)-rounded neurospheres with entrapped MCs (shown in Fig. 7B) had an average length and width of 939±290 μm and 723±200 μm. qRT-PCR analysis of mRNA transcripts of these neurospheres showed upregulation of neural progenitor markers, PAX6, SOX1, Nestin, and MSI, and the neuronal marker, MAP2, as compared to undifferentiated cells (Fig. 7A). Upregulation of the endoderm marker AFP and mesoderm marker HAND1 indicated the possible presence of cells from the endodermal and mesodermal lineages, similar to that observed in the 2D and static MC cultures (Fig. 5I). Neural progenitor markers Nestin and SOX1 (not shown) were also observed in MC neurospheres by immunohistological staining (Fig. 7C). The plated dissociated single cells from the neurospheres were also stained positive for 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 7D) and PAX6 (Fig. 7E) and Nestin (Fig. 7F).

Characterization of iPS (IMR90) spinner MC culture-derived neurospheres.
Similar results were obtained with the HES-3 MC-based neurospheres. NPC-related gene expressions were upregulated as shown in Figure 8A. HES-3-derived neurospheres had a similar culture morphology as iPS (IMR90) ones (Fig. 8B); the cell–MC neurosphere was also stained positive for Nestin NPC marker (Fig. 8C), and single cells obtained from the neurospheres stained positive for DAPI, PAX6, and Nestin (Fig. 8D–F).

Characterization of HES-3 spinner MC culture-derived neurospheres.
To test the potency of these NPCs to generate the three neural lineages, the neurospheres from day-18 or day-20 cultures were plated onto a laminin-coated TC plate and were allowed to differentiate for another 14 days. After this, cells were stained positive for the neuronal marker, βIII-tubulin (Fig. 9A), the astrocyte marker, GFAP (Fig. 9B), and the oligodendrocyte marker, O4 (Fig. 9C). Furthermore, after BDNF-, GDNF-, and NGF-based directed neuronal differentiation, patch-clamp recordings of differentiated neurons from NPCs showed spontaneous postsynaptic currents (black) and membrane potential recordings (red); two representative examples are shown in Figure 9D.

Nondirected and directed differentiation of NPCs. Nondirected differentiation was done by plating neurospheres on a laminin-coated plate. Cultures were positively stained for the neuronal marker
Discussion
hESCs and hiPSCs are known to have similar phenotypes; they are characterized by the same pluripotent markers, have the ability to differentiate into the 3 germ layers, and display normal karyotypes. Thus, it was not surprising that they have similar growth kinetic properties. In static MC cultures, HES-3 and iPS (IMR90) cell lines propagated in the mTeSR1 serum-free defined medium achieved cell densities of 1.2–1.5×106 cells/mL, 8–15-fold cell expansion, and doubling times of 36–42 h (Table 1 and Chen et al. 30 ). Moreover, they show similar metabolic rates. Specific consumption of glucose and glutamine are 0.302–0.444 and 0.034–0.046 mmol/109 cells/h, respectively, and lactate and ammonium production rates are 0.555–0.801 and 0.015–0.018 mmol/109 cells/h, respectively (unpublished results and Chen et al. 30 ).
MC cultures were used by several groups for expansion of several lines of hESCs.18–24,30,31 However, there is only one report of expansion of hiPSCs in MC cultures described in a review article by Kehoe et al. 22 Earlier, our group developed an MC platform for hESC expansion.18,30,31 A variety of MCs having different shapes, surface characteristics, and sizes had been tested for propagation of different hESC lines in serum-free and serum-containing media.30,31 We found that to achieve pluripotent hESC growth for multiple passages, the MCs should be coated with ECM proteins (Matrigel, laminin, or vitronectin).30,37 We also showed that agitation of hESCs in spinner cultures could cause spontaneous differentiation of hESCs, 27 presumably as a result of mechanical stress.
In this study, we extended the utility of this platform and presented a scalable stirred flask system to expand iPS (IMR90) cells. Matrigel-coated, cylindrical-shaped, cellulose DE-53 MCs having a diameter of 35±7 μm and length of 130±60 μm were chosen for this study, since they were found to allow very efficient cell attachment (over 90% in 2 h), to support pluripotent hESC growth for multiple passages, and generate compact cell–MC aggregates probably due to their elongated cylindrical structure.18,31
We indeed demonstrated that iPS (IMR90) cells can also be propagated in static MC cultures for many passages, expressing pluripotent markers on these Matrigel-coated cylindrical MCs achieving cell yields of about 1.2×106 cells/mL, similar to the yields reported previously by Chen et al., 2010, 30 for HES-3 cells.
Propagating iPS (IMR90) cells in a stirred spinner flask, MC cultures yielded a high cell density of about 3×106 cells/mL probably as a result of better mass transfer obtained with the smaller aggregate size achieved in the stirred culture. These yields are similar to the ones obtained with HES-3 cells (Table 1). However, a small secondary peak seen in the histograms of mAb84 and TRA-1-60 markers indicated the presence of a small differentiating population of cells. This phenomenon was also seen in HES-3 MC culture, and it was attributed to either the mechanical stress as a result of agitation conditions 27 or the effect of low pH and/or high lactate concentration at the last stage of cell growth. 30 Chen et al., 2010, 30 reported that lactate concentration >1 g/L resulted in slower growth of hESCs and the downregulation of pluripotent markers.
Changing the feeding regime to twice-daily medium exchange leads to an increase in cell expansion by twofold over once-daily medium exchange. This feeding regime also reduced the presence of spontaneously differentiating populations of cells achieving expression of pluripotent marker (OCT3/4, mAb84, and TRA-1-60) levels similar to static 2D cultures. This phenomenon, which was also shown in hESC cultures (results not shown), indicates that the differentiating effect of the mechanical stress can be overcome by changing the metabolic conditions of the cultures. The metabolic conditions can be changed by lowering the concentration of the waste metabolite (i.e., lactate), increasing the concentration of unknown nutrients and growth factors, or controlling environmental conditions (e.g., pH). Further studies are needed to clarify these issues. With a twice-daily medium exchange and a seeding density of 3×105 cells/mL, our system achieved 20-fold pluripotent stem cell expansion and a cell density of 6×106 cells/mL. These cell yields are higher than those achieved in all previously reported expansion systems of hiPSCs and hESCs.
There are only a few reports on the integrated expansion and differentiation of hESCs in MC cultures.21,26,28 Integration of expansion and differentiation in one continuous spinner culture can be problematic, since the expansion conditions are not optimal for differentiation, and/or the differentiation protocol requires manual handling, which cannot be done in stirred cultures. Serra et al. 29 and Steiner et al. 28 both reported integrated expansion and directed differentiation systems of hESC aggregates. Both systems achieved only 2-5-fold expansion per passage. Steiner et al., 2010, 28 further differentiated NPCs to neurons, oligodendrocytes, and astrocytes, but the yield of NPCs achieved was not studied.
The classical 2D neuroprogenitor differentiation protocol requires several medium exchange steps and a series of EB formation, cell selection, and plating steps 33 (Fig. 1A). We were able to completely eliminate the replating and selection steps. The MC–cell aggregates obtained during the expansion phase were further differentiated to neurospheres simply by medium changes (Fig. 1B, C). In the first part of the study, the new differentiation protocol was tested in static MCs in six-well plates (Fig. 1B) and compared to the classical 2D protocol 33 (Fig. 1A). Undoubtedly, the 2D differentiation protocol has the advantage of selection of colonies that were richer in NPCs. However, even without this labor-intensive colony selection, our MC differentiation method yielded a similar density of PSA-NCAM+ NPCs. Volumetric NPC yield was 2.2±0.47×106 NPCs/mL in MC culture, as compared with 2.3±0.14×106 in the 2D condition (Table 1).
In the second part of this study, we combined the 7-day twice-daily feeding cell expansion followed by an 18-day differentiation in spinner flasks. This resulted in a production of 333 PSA-NCAM+ NPCs generated from every iPS (IMR90) cell seeded in MC spinner culture as compared to 53 PSA-NCAM+ NPCs/iPS (IMR90) in 2D differentiation cultures. It is important to note that at day 20, there is a drop in the cell density in the spinner flask culture, probably due to the inability of the current medium conditions to sustain the very high cell densities of 10.6×106/mL, probably due to some limiting nutrient. Thus, there is still room for further improvement in the bioprocess by additional medium feeding.
The universality of the integrated expansion differentiation process was demonstrated by applying it to hESC HES-3. Similar high yields of 371 PSA-NCAM+ NPCs per seeded hESC were achieved. NPCs are an excellent starting source of cells for further differentiating into neurons, oligodendrocytes, and astrocytes useful for cell therapy or for drug-screening applications. By developing a robust high-yielding process for generating NPCs, these cells could become a uniform source for screening small molecules for neuronal differentiation,38,39 studying neural development in neurodegenerative diseases such as amyotrophic lateral sclerosis, 14 familial dysautonomia, 15 SMA, 16 and Rett syndrome, 17 or further directed differentiation to important lineages such as neurons for treatment of Parkinson's disease. NPCs can also be used to study bioactive surfaces and scaffolds for neural differentiation in tissue-engineering applications.40,41
In conclusion, we have demonstrated a superior MC-based platform for pluripotent stem cell expansion and differentiation to NPCs in suspension cultures. The MC platform is robust, and the NPCs generated are 6-fold to 11.6-fold higher than the conventional 2D platform. It is our long-term goal to further develop this MC-based NPC differentiation bioprocess, to produce specific neural cell types such as neurons, oligodendrocytes, or astrocytes to meet the demands of these cells for cell therapy, disease studies, drug screening, and tissue engineering.
Footnotes
Disclosure Statement
No competing financial interests exist.