
Abstract
Human mesenchymal stem cells (hMSCs) that can differentiate into chondrocytes are a potential autologous cell source for repair of damaged tissue. Current methods usually induce the formation of all three chondrocyte phenotypes, hyaline, fibrous, and elastic, without the ability to selectively induce only one of them. By controlling the size of hMSC cell clusters, it may be possible to direct differentiation more uniformly toward hyaline chondrocytes. We designed new cell culture platforms containing microwells of different diameters. The platforms and wells were composed of a zirconia ceramics substratum. hMSCs briefly adhered to the substratum before releasing and entering the microwells. The physical restraints imposed by the microwells enabled hMSC clusters to homogenously differentiate into hyaline chondrocyte-like cells. Chondrogenic aggregates in microwells expressed the hyaline chondrocyte-specific genes Col II, aggrecan (ACAN), and cartilage oligomeric protein (COMP). The cultures also produced hyaline chondrocyte-specific matrix proteins Col II and ACAN homogenously throughout the aggregates. In contrast, chondrogenesis in pellet cultures was heterogeneous with the expression of nonhyaline chondrocyte genes CD105, Col X, and Col I. In these pellet cultures, hyaline and nonhyaline chondrocyte-specific matrix proteins were distributed heterogeneously. Thus, this novel ceramic microwell substratum technology efficiently directed the differentiation of hyaline chondrocyte-like cells from hMSCs. These results indicate that there is a close relationship between hMSC cluster size regulation in the microwells and differentiation tendency. This microwell culture differentiation method will provide a valuable experimental system for both experimental and potential clinical studies.
Introduction
Human mesenchymal stem cells (hMSCs) can be isolated easily from various tissues using standardized methods and are expected to be a practical cell source for regenerative medicine. 4 Several techniques, such as the pellet method and micro-mass cultures, utilize the transforming growth factor-β (TGF-β) family of cytokines to promote the differentiation of hMSCs into chondrocytes.5,6 Other methods include the use of scaffolds to produce chondrogenic constructs of larger sizes. Using these methods, hMSC pellets induced with TGF-β3 synthesize cartilage matrices containing type II collagen and proteoglycan and form hyaline cartilage-like tissue.7,8 However, these methods also induce the expression of type X collagen, which is an early marker of hypertrophic chondrocytes and is normally absent from hyaline cartilage. 9 Thus, certain other factors required to produce high-quality hyaline cartilage appear to be missing from current induction protocols.
Cellular differentiation is influenced by cell-autonomous programs, soluble factors, and physical forces, as well as the surrounding three-dimensional (3D) architecture. 10 Therefore, 3D cultures have been tested to provide cultured cells with a microenvironment that is similar the natural in vivo microenvironment.11–13
The control of cell cluster size is important because differentiation is affected by cell number and cell-to-cell contact. 14 Culture chips with multiple cavities in a triangular arrangement can control the diameter of cell clusters produced from a liver-derived cell line. 15 The adhesion surfaces of these chips are coated by a low-attachment material to facilitate the formation of the cell clusters. Consequently, the clusters that form do not adhere to the bottom of the culture chip cavities. Cell culture using low-attachment dishes is unsuitable for long-term culture because it is difficult to replace the culture medium. Furthermore, because differentiation depends, in part, upon cell attachment, such culture techniques do not promote differentiation. 16
We therefore decided to study the feasibility of chondrogenic differentiation for MSC clusters by using a new 3D culture substratum that can control the cell cluster size while promoting adherence of the cell clusters. Ceramics such as zirconia, alumina, titania, and hydroxyapatite are inorganic and nonmetallic solid materials prepared by the application of heat and subsequent cooling. They are used in the manufacture of subframes for the construction of dental, bone, and articular restorations. Besides, there are several studies in the literature in which ceramics, such as hydroxyapatite and zirconia, have been used for the differentiation of MSC and might play a role in promoting a specific cell lineage differentiation.17,18 We designed a ceramics platform containing microwells arranged in a grid pattern. To obtain uniform cell clusters in the microwells, we examined the optimum raw ceramic materials for seeding to establish clusters within the microwells. When cultured on the microwell substratum, the hMSC clusters efficiently underwent hyaline chondrogenic differentiation.
Materials and Methods
Preparation of ceramic microwell substratum
The ceramic microwell substratum (CERAHIVE™; Covalent Materials Co., Ltd., Kanagawa, Japan) was prepared by microfabrication technology. First, a slurry was prepared by ball milling zirconia powder HSY3.0 (Daiichi Kigenso Kagaku Kogyo Co., Ltd., Osaka, Japan) with distilled water and 8% (w/w) polycarboxylate (Toagosei Co., Ltd., Tokyo, Japan) for 3 h. Next, 5% (w/w) polyethyleneimine (BASF Japan Co., Ltd., Tokyo, Japan) and 0.8% (w/w) sorbitol polyglycidyl ether (Nagase ChemteX Co., Ltd., Osaka, Japan) were added to the slurry and stirred. The mixture was cast by pouring it into a microfabricated mold. The ceramics gels were removed from the mold, dried, and sintered for 2 h at 1150°C or 1600°C (Fig. 1). The substrata were imaged by scanning electron microscopy (FE-SEM [S-4800]; Hitachi High-Technologies Co., Ltd., Tokyo, Japan].

hMSC culture
UE6E7T-2 cells prepared from a bone marrow-derived immortalized hMSC line19,20 were obtained from RIKEN BioResource Center (Ibaraki, Japan). They were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum. When the cells reached confluence, they were passaged using 0.25% trypsin/EDTA. Cultures were maintained in a humid atmosphere of 5% CO2 at 37°C.
Chondrogenic differentiation in pellet culture
hMSCs (2.5×105) were placed in 15-mL conical polypropylene tubes and centrifuged at 500 g for 5 min at 20°C. The cells were resuspended in chondrogenic culture medium consisting of high-glucose DMEM containing 100 μg/mL sodium pyruvate (Life Technologies Japan Ltd., Tokyo, Japan), 10 ng/mL TGF-β3 (R&D Systems, Minneapolis, MN), 100 nM dexamethasone (Sigma, St. Louis, MO), 1% insulin–transferrin–selenium (ScienCell Research Laboratories, Carlsbad, CA), 40 μg/mL proline (Sigma), and 0.1 mM ascorbic acid (Sigma). 21 The suspensions were re-centrifuged to form cell pellets at the bottom of the 15-mL tubes. The pellets were maintained with 1.0 mL of chondrogenic culture medium in a humid atmosphere of 5% CO2 at 37°C. The medium was replaced every fourth day for up to 21 days.
Chondrogenic differentiation in the microwell substratum
Confluent hMSCs grown on 10-cm dishes were released by trypsin/EDTA as described above. The cells were seeded in 100 μL of medium onto microwells set in 24-well plates. The number of cells seeded was increased according to the diameter of the microwells: 1×105 hMSCs in 100-μm microwells (2741 microwells), 2×105 hMSCs in 200-μm microwells (973 microwells), 4×105 hMSCs in 400-μm microwells (285 microwells), and 8×105 hMSCs in 600-μm microwells (137 microwells). After 6 h, 700 μL of culture medium was added to the microwell substratum. After 4 days of incubation, the hMSC culture medium was aspirated and replaced by 800 μL of chondrogenic culture medium. Culturing continued in a humid atmosphere of 5% CO2 at 37°C, and the medium was changed every fourth day for up to 21 days. The MSC aggregates and the chondrocyte-like aggregates that had formed in the microwells were fixed in 2.5% glutaraldehyde in phosphate-buffered saline (PBS) and 1% osmium tetroxide in PBS. After rinsing and dehydration in sequentially increasing concentrations of ethanol solutions (up to 100%), the dehydrated samples were immersed in t-butyl alcohol and finally dried in a freeze-dry system (Labconco, Kansas City, MO). After sputter coating with platinum, the substrata and MSC aggregates were imaged by FE-SEM. For glycosaminoglycan (GAG) and DNA measurement, reverse transcription–polymerase chain reaction (RT-PCR), and immunohistochemistry, all cell aggregates in one substratum were collected by pipetting.
GAG and DNA measurement
Chondrocyte-like aggregates in the microwells and chondrocyte-like pellets in 15-mL tubes were harvested at 7, 14, and 21 days for GAG and DNA quantification. GAGs were measured with the Acidic Mucopolysaccharide Assay Kit (Primary Cell, Hokkaido, Japan), 22 and the DNA quantitation was carried out with the DNeasy Blood & Tissue Kit (Qiagen K.K., Tokyo, Japan) 23 and the PicoGreen dsDNA Quantitaion Kit (Molecular Probes, Eugene, OR) with phage l DNA as standard.
Gene expression analysis
mRNA levels were analyzed for genes typically expressed by hMSCs and the different chondrocyte phenotypes (Table 1). In addition to the pellet and microwell aggregates, we also analyzed the gene expression of normal human hyaline chondrocytes at passage 1 (Lonza Walkersville, Inc., Walkersville, MD). The cells were thawed, seeded, and cultured for 12 h using the CGM BulletKit (Lonza Walkersville, Inc.).
hMSC, human mesenchymal stem cells; TGF, transforming growth factor.
For all cultures, total RNA was extracted using NucleoSpin RNA II (Takara Bio Corp., Shiga, Japan) and reverse transcribed using the Quantitect Whole Transcriptome Kit (Qiagen K.K.). The expression of CD29, CD44, CD105, Col I, Col II, Col X, aggrecan (ACAN), cartilage oligomeric protein (COMP), SOX9, Runx2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was assessed in a Thermal Cycler Dice Real Time System II (Takara Bio Corp.) using the primers listed in Table 1. PCR was performed with Takara Ex-Taq (Takara Bio Corp.) using a 10-min incubation at 94°C followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and a 5-min incubation at 72°C after completion of the final cycle. The amplification products were analyzed by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
Real-time quantitative RT-PCR was performed for Col II, ACAN, and Col X. Using primers from Takara Bio Corp., the reactions were performed in a Thermal Cycler Dice Real Time System II (Takara Bio Corp.) with SYBR Premix Ex Taq II (Takara Bio Corp.). The relative expression levels for each primer set were normalized to the expression of GAPDH, following the manufacturer's protocol. 24 The gene expression changes were expressed as fold changes. For all genes evaluated, the fold change was calculated by dividing the value for each sample by the value for the pellet method at day 7.
Histology and immunohistochemistry
The chondrocyte-like aggregates produced in pellet and microwell cultures were fixed in 4% paraformaldehyde for 1 h and then embedded in paraffin. Serial sections (5-μm-thick) were deparaffinized and rehydrated through a series of graded ethanol solutions and then stained with 0.1% safranin O and 0.1% Fast Green solution (Wako Pure Chemical Industries, Ltd., Osaka, Japan). The sections were imaged using light microscopy (VHX-500; Keyence Corporation, Osaka, Japan).
The sections were singly or doubly stained with antibodies for Col II (1:100 rabbit polyclonal; AbD Serotec, Kidlington, Oxford, United Kingdom), Col I (1:100 rabbit polyclonal; Abcam, Cambridge, Cambridgeshire, United Kingdom), Col X (1:100 rabbit; LSL Co., Ltd., Tokyo, Japan), ACAN (1:25 mouse monoclonal; AbD Serotec), CD105 (1:50 mouse monoclonal; Abcam), elastin (1:100 rabbit; LSL Co., Ltd.), and Ki67 (1:50 rabbit; Santa Cruz, Inc., Santa Cruz, CA). After primary antibody incubation, the samples were incubated with a mixture of appropriate secondary antibodies conjugated with Alexa Fluor 488 or 568 and 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) to stain the nuclei (Molecular Probes). The specimens were observed with a Leica TCS SP2 AOBS spectral confocal laser scanning microscope equipped with Ar, He/Ne, and blue diode lasers.
Apoptosis assay
The sections were processed for TUNEL using the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Millipore, Billerica, MA) according to the manufacturer's instructions. DNA fragments labeled with digoxigenin nucleotide were detected using a sheep anti-digoxigenin antibody conjugated with FITC as supplied in the assay kit.
Statistical analysis
All experiments were performed at least 3 times, and the data were expressed as mean±standard deviation. For comparisons between groups, t-tests were performed using Minitab Statistical Software (State College, PA). In all analyses, p<0.05 was considered significant.
Results
Formation of hMSC clusters in the zirconia microwell substrate
hMSC morphology was examined on zirconia microwells sintered at 1150°C to create a porous structure and at 1600°C to create a dense structure (Fig. 2A). For the porous substrate (Fig. 2A), the hMSCs adhered to the surface, but not within the microwells (Fig. 2B), by the day following inoculation. Thereafter, the cells gradually detached from the surface, and by days 2–3 of culture, some of the cells became trapped in the microwells (Fig. 2B). Subsequently, the trapped hMSCs self-assembled to form cell clusters that adhered to the bottom of the microwells within 2–4 days of culture. By day 2 of culture, the DNA content increased by 1.4-fold (Fig. 2C), but beyond 3 days, the DNA content decreased. This change could reflect a balance between the loss of detached cells and the proliferation of cells within the microwells. In contrast, when the cells were seeded onto the dense substrate (Fig. 2A), they formed confluent monolayers without self-assembling into cell clusters. In addition, hMSCs were seeded on different types of microwell substrata such as hydroxyapatite, titania, and alumina, but they did not form cell clusters (Supplementary Fig. S1; Supplementary Data are available online at

To determine diameters of the cell clusters formed in microwells of different sizes, we seeded 1×105, 2×105, 4×105, and 8×105 hMSCs on the porous substrate in microwells with diameters of 100, 200, 400, and 600 μm, respectively (Fig. 2A). After 4 days of culture, the hMSCs formed clusters with sizes and shapes that depended on the dimensions of the microwells. The mean diameter of the cell clusters in the microwells was 65±5.3, 120±11.7, 240±19.8, and 360±42.0 μm, respectively.
Chondrogenic differentiation of hMSCs in microwells
To investigate the effects on hMSC differentiation of the physical restraints imposed by the microwells, hMSCs were seeded on the microwell substrata and cultured in a chondrogenic culture medium. For each microwell size, the induced clusters remained attached to the bottom of the microwells and produced components that were similar to pellet surface induced by pellet culture (Fig. 3A). For microwells with diameters of 400 μm or less, the induced chondrocyte-like clusters were confluent and therefore could not grow further because of the physical constraints of the microwell surfaces. In the microwells with diameters of 600 μm, the cell clusters were not confluent and thus had room to grow and become hypertrophic.

Next, we assessed the mRNA expression of CD105 (a marker for hMSCs), Col II (a marker for hyaline chondrocytes), and Col X (a marker of hypertrophic chondrocytes). For microwells with diameters of 400 μm or less, the induced hMSC clusters expressed Col II but not CD105 or Col X (Fig. 3B). In contrast, cell clusters formed in the 600-μm diameter microwells expressed CD105, Col II, and Col X.
The distribution of the hypertrophic chondrocyte marker Col X among cells in the clusters was examined by immunohistochemistry. We could not obtain serial sections of aggregates formed in microwells with diameters of 100 or 200 μm, because they were too small. Therefore, we used chondrocyte-like aggregates in the 400-μm microwells to represent aggregates grown in microwells with small diameters. Col X was not detected in the matrix of aggregates cultured in the 400-μm microwells (Fig. 3C); however, it was distributed near the surface of the cell aggregates cultured in the 600-μm microwells.
Proteoglycan production in pellet and microwell cultures
The assembly of cartilage matrices in microwell and pellet cultures was assessed by determining the GAG content. The accumulation of GAGs in pellet cultures was rapid up to 14 days but much slower from 14 to 21 days (Fig. 4A). In contrast, GAGs in the microwell cultures increased gradually up to 21 days. After 21 days, the amount of GAGs in pellet cultures was 2.5-fold higher than that in microwell cultures. Based on the DNA content, the number of chondrocyte-like cells in the pellet cultures was fivefold higher than that in the microwell cultures. In the pellet cultures, the ratio of GAGs to DNA increased up to 14 days and then remained relatively constant up to 21 days. For cells in microwell cultures, the ratio increased linearly over 21 days.
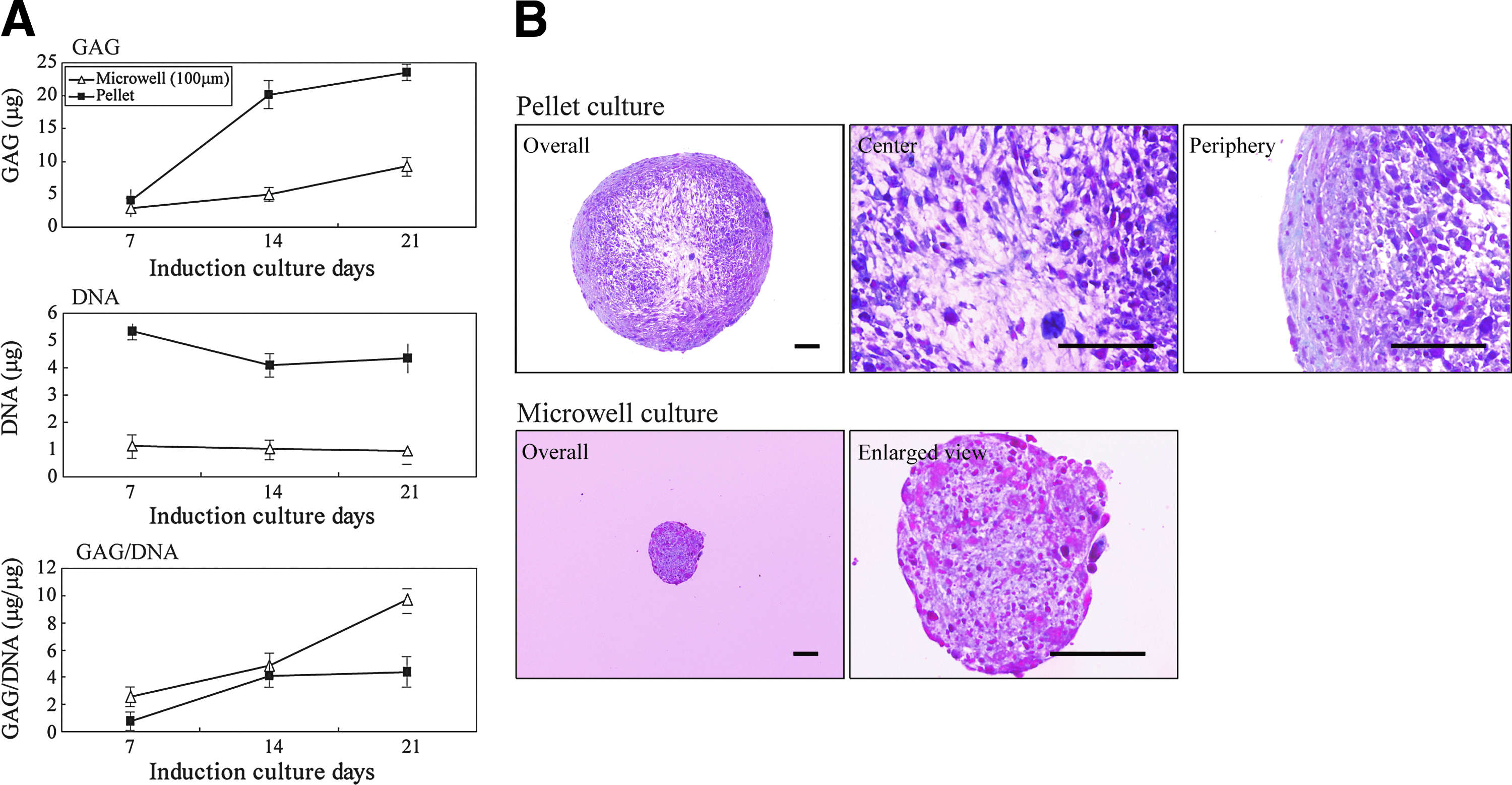
Comparison of DNA content and glycosaminoglycans (GAG) accumulation between one pellet in pellet culture and all cell aggregates harvested from one substratum in microwell (100 μm) culture.
We examined the morphological distribution of the matrix components of chondrocyte-like cells induced in pellet and microwell cultures for 21 days by staining with safranin O and Fast Green (Fig. 4B). For aggregates in pellet culture, the stained polyanions associated with the cartilage matrix were distributed mostly in the peripheral region of the clusters, with only a minimal amount in the center. In contrast, for microwell cultures, the safranin O staining was uniformly distributed throughout the aggregates.
Time course of the expression of cartilage-specific genes in pellet and microwell cultures
We examined differences in gene expression between pellet and microwell cultures for various cartilage macromolecules, transcription factors, and extracellular matrix components (Fig. 5). The gene expression of chondrocytes derived from human hyaline cartilage was evaluated with RT-PCR assays. On day 21 of culture, these cells expressed CD29, CD44, COMP, Col II, ACAN, and SOX9, but not Col X, Col I, or Runx2. Gene expression of CD29 and CD44 was shared by undifferentiated hMSC and hyaline cartilage lineage. To determine the temporal expression pattern during differentiation to chondrocyte-like cells, hMSC aggregates were probed by RT-PCR for cartilage-specific gene expression on days 7, 14, and 21. In pellet culture, the typical hMSC marker genes CD29, CD44, and CD105 were expressed throughout the culture period. The expression of the chondrocyte marker genes Col II, SOX9, and Col X increased progressively, whereas that for COMP and ACAN remained steady from day 7 to 21. In contrast, for aggregates grown in microwell cultures, the expression of CD29 and CD44 was steady for 21 days, but CD105 expression was not detected. The expression of COMP, Col II, ACAN, and SOX9 increased gradually. On day 7, the expression of Col X was barely detectable, and that of Col I was undetected.
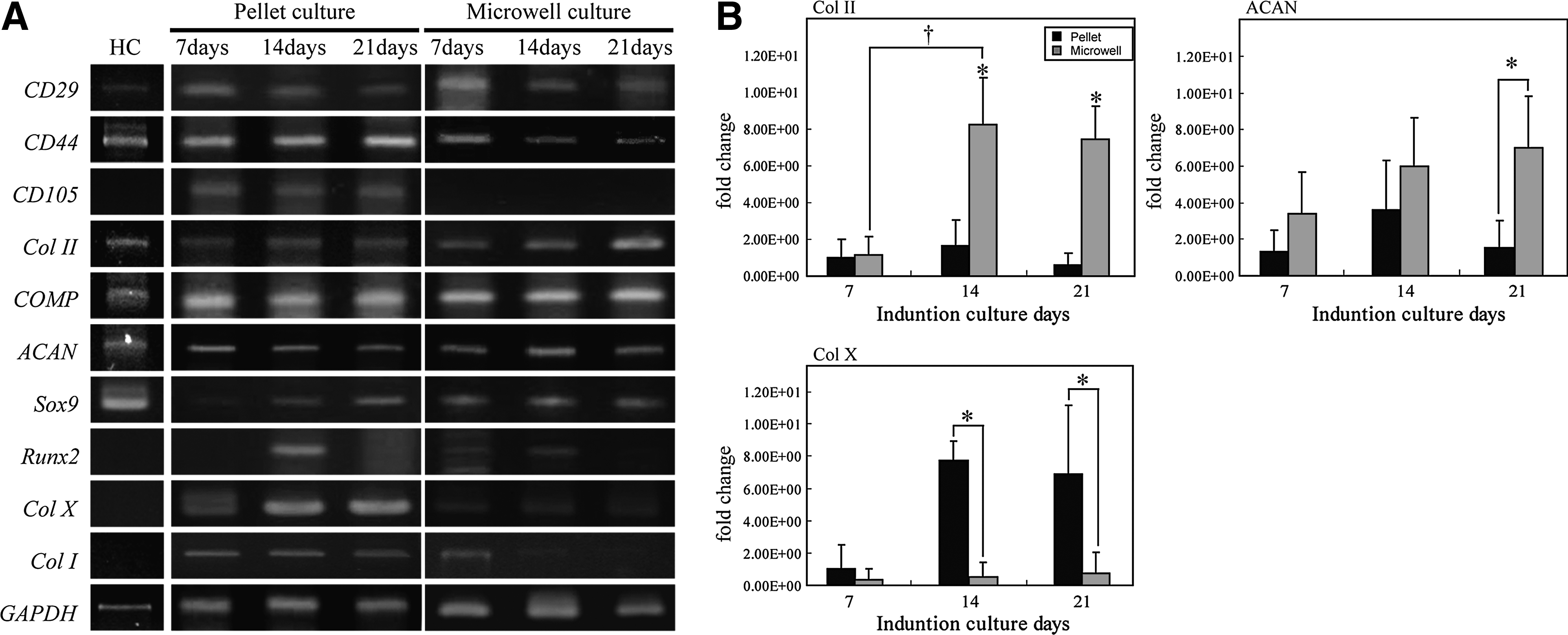
Finally, the expression of the major chondrocyte-specific genes Col II, ACAN, and Col X was determined by quantitative RT-PCR. The expression of Col II in pellet cultures increased by 2-fold between days 7 and 14 and then decreased slightly on day 21 (Fig. 5B). In contrast, the expression of Col II in microwell cultures increased 8-fold between days 7 and 14 and was then maintained at that level to day 21. On days 14 and 21, the expression of Col II in the microwell cultures was approximately 5-fold and 12-fold greater, respectively, than that of pellet cultures. In pellet cultures, the expression of ACAN increased 2-fold between days 7 and 14 and then decreased by half between days 14 and 21 (Fig. 5B). In microwell cultures, the expression of ACAN increased 2.8-fold between days 7 and 21. The expression of ACAN in microwell cultures was approximately 3-fold higher than that in pellet cultures on day 21. The expression of Col X in pellet cultures increased 7-fold between days 7 and 14 and then remained stable until day 21 (Fig. 5B). However, in microwell cultures, the expression of Col X was low throughout the culture period, reaching only 0.06–0.1 times that of the level in pellet cultures on days 14 and 21, respectively.
Localization of extracellular matrix molecules and cell surface antigens
Immunolocalization was used to assess the morphological distribution of matrix components within the differentiated microwell and pellet cultures at day 21 (Fig. 6). In microwell cultures, Col II and ACAN were uniformly distributed throughout the induced aggregates (Fig. 6A). In pellet cultures, the distribution of Col II, ACAN, and CD105 was heterogeneous. Col II and ACAN were distributed peripherally, and CD105 demonstrated distinct immunoreactivity at the center of the pellet. The staining of Col X was weak throughout the chondrogenic tissue in microwell cultures, but it was distributed at the surface of pellet culture aggregates (Fig. 6B). Col I was minimally expressed in all regions of the aggregates obtained by microwell culture. In pellet cultures, Col I staining was distributed at the surface and at the central region of the aggregates. Elastin was not detected in aggregates induced by microwell culture, but it was detectable at the surface of pellet culture aggregates (Fig. 6B). Hence, the microwell method induced hMSC aggregates to form chondrocyte-like cells with a more homogeneous distribution of matrix components when compared with the pellet method.

Comparison of matrix synthesis and apoptosis induced cells in chondrocyte aggregates induced in microwell (400 μm) and pellet cultures.
In 21-day cultures, TUNEL-positive cells and Ki67-positive cells were detected to identify whether the cells underwent apoptosis or proliferated (Fig. 6B). In microwell cultures, apoptotic cells were dispersed throughout the aggregated chondrocyte-like cells. In contrast, in pellet cultures, apoptotic cells were localized mostly in the central area, with few observed in the peripheral margin of the aggregates. Proliferating cells were scattered throughout regions of the aggregates obtained by the microwell and pellet culture methods. In addition, double staining of TUNEL and Col II/Col I/CD105 was used to identify the type of cells that underwent apoptosis (Supplementary Fig. S2). In the pellet cultures, some Col II-, Col I-, and CD105-positive cells underwent apoptosis. In contrast, in the microwell cultures, some Col II- and Col I-positive cells underwent apoptosis, but CD105-positive cells were not observed.
Discussion
Recent investigations have employed 3D scaffolds generated from materials such as collagen, silk, hydrogels, and porous membrane supports to produce cartilaginous constructs of the regeneration of large cartilage defects,25–27 but little is known about the effectiveness of cell differentiation in such scaffolds. Generically, the preparation of culture plates for controlled cell clustering is a complicated process that requires the definition of areas of cell adhesion and nonadhesion using microarray technology. 28 The use of microwells for culture does not require the immobilization of cell clusters at defined locations or the control of cluster diameter. Therefore, we tested the effect of 3D microwell culture using a ceramic-based substrate on the chondrogenic differentiation of hMSCs.
First, we assessed the behavior of the hMSCs in the microwells. The hMSCs behaved differently on microfabricated structures composed of submicron-sized zirconia particles when compared with hMSCs cultured on dense zirconia structures composed of micron-sized particles. After a brief attachment over 1–2 days to the porous surface, the cells detached and accumulated within the microwells. It is likely that this plating process by itself might preferentially select a subpopulation of the MSCs that can attach to the bottom of porous zirconia microwells and happen to possess more homogenous chondrogenic potential. Thus, the adhesion between the isolated cells and the microfabricated porous surfaces was weak. This low level of adhesion between the cells and the ceramic surfaces promoted the formation of cell clusters in the microwells. It is conceivable that the area of contact between the cells and the ceramic particles affects the cell-to-material adhesion force and cell migration to provide cell cluster formation within the microwells. However, it has also been reported that thin films coated with nano-sized zirconia particles enable hMSCs to adhere and proliferate. 29 Furthermore, in the present study, hMSCs did not form clusters on different microwell substrate materials such as hydroxyapatite. It is therefore likely that there are optimal particle sizes and raw ceramic materials that enable hMSCs to form clusters. Such particle sizes and material morphologies may be obtained using principles such as micro-imprinting 30 and nano-pillars. 31 In culture chips, 17 the mean diameter of the cell clusters in microwells depended on the density of inoculated cells, and the diameter of the cell clusters increased with an increase in the distance between microwells on the chip. These findings are similar to those obtained with the microwell substrate in the present study.
The commonly used pellet culture method for chondrogenic differentiation of hMSCs is effective. In the present study, we observed chondrogenic differentiation in microwells that was similar to the differentiation observed in pellet cultures. There were, however, some differences in chondrogenic features between the pellet and microwell cultures. Pellets of hMSCs stimulated with TGF-β3 and dexamethasone expressed the hyaline cartilage–specific genes Col II and ACAN. 32 They also expressed chondrogenic genes not normally expressed in hyaline cartilage, such as Col X (an early marker of the hypertrophic chondrocytes), 33 and Col I (a marker of dedifferentiation and/or a marker of fibrous cartilage).34,35 Expression of the hMSC marker CD105 was also maintained for 3 weeks. Thus, the cells cultured using the pellet method were composed of not only the hyaline chondrocyte-like phenotype but also the hypertrophic chondrocyte-like, fibrous chondrocyte-like, and elastic chondrocyte-like phenotypes, as well as undifferentiated cells. Immunostaining and safranin O staining of pellet cultures revealed that chondrogenic components such as GAGs were distributed in the periphery of the clusters, with sparse distribution in the central regions. Col II, Col X, and elastin in the pellet aggregates were distributed heterogeneously at the peripheral margin but were not detected in the central region of the pellets. In addition, CD105 was distributed in the central region. Thus the chondrocyte-like cells induced in the pellet culture were heterogeneous and mainly distributed in the peripheral regions of the clusters, whereas undifferentiated hMSCs were located in the central regions. The induction of nonuniform chondrocyte-like cell aggregates by the pellet method has been reported,36,37 but with the exception of cells expressing Col X, the appearance of various chondrocyte-like phenotypes has not been documented. More studies are necessary to determine factors that affect the differentiation of hMSCs to enhance the production of hyaline chondrocytes.
Clusters of hMSC-derived cells in the microwell substrata induced by TGF-β3 and dexamethasone expressed hyaline cartilage genes Col II, ACAN, and COMP. Simultaneously, very little of the other chondrocyte marker genes, Col I, Col X, and the undifferentiated hMSC marker CD105 were expressed. The hyaline chondrogenic matrix components Col II and ACAN were distributed homogeneously in the microwell cultures, which expressed minimal amounts of markers that are not typically expressed in hyaline cartilage, that is, Col I, Col X, and elastin. These observations suggest that the phenotype induced by the microwell method is homogeneous and is composed of hyaline chondrocyte-like cells. Moreover, the chondrocyte-like cells induced by the microwell culture synthesized more GAGs per cell for 21 days than cells in the pellet culture. Thus, the elevated GAG synthesis and hyaline chondrocyte gene expression in cells on the microwell substrate suggests a greater efficiency of hyaline chondrocyte-like cell formation from hMSCs when compared with the pellet cultures. The amount of DNA in the chondrogenic aggregates in the microwell cultures and pellet cultures did not change over 21 days of culture. However, the number of TUNEL-positive cells was higher than the number of Ki67-positive cells after 21 days of culture. Therefore, we consider that the number of hyaline chondrocyte-like cells in the aggregates gradually decreased over a longer culture period.
Compared with fibrous and elastic cartilage, hyaline cartilage plays distinct roles. Hyaline chondrocytes also form articular cartilage as a permanent tissue. 38 To obtain hyaline cartilage from stem cells such as hMSCs, it is essential to induce the formation of hyaline chondrocytes without further differentiation into hypertrophic chondrocytes. Although the pellet culture method effectively induces the formation of chondrocyte-like cells, the efficient production of hyaline chondrocyte-like cells is reduced by the formation of other types of chondrocyte-like cells.
Many studies have reported that differentiation is influenced by several key factors, including the presence of appropriate growth factors, 39 low oxygen tension, 40 high pressure, 41 and the formation of 3D cell clusters. 42 The focus of those studies was not on the selective induction of hyaline chondrocytes. Some factors that influence differentiation are known. For instance, bone morphogenetic protein 2 (BMP-2) and dexamethasone can inhibit the expression of Col X and maintain the cells in a nonhypertrophic status. 43 Similarly, parathyroid hormone-related protein (PTHrP) can inhibit hypertrophy. 44 However, the in vitro regulation of hyaline chondrocyte hypertrophy remains unclear, and these factors cannot prevent differentiation to other chondrocyte phenotypes. Therefore, the chondrocytes induced by these protocols are considered heterogeneous. A principal finding of our study was that small microwells are effective for inducing hMSCs to form hyaline chondrocyte-like cells. Even without the addition of BMP-2 and PTHrP, this new methodology is efficient and does not produce hypertrophic, elastic, or fibrous chondrocyte-like cells.
We investigated the effect of physical constraints imposed by microwell size on differentiation. hMSCs in microwells with diameters of 100, 200, 400, and 600 μm were induced to form chondrocyte-like cells with TGF-β3 and dexamethasone. The hMSC clusters in the largest microwells (600 μm) tended have higher expression of Col X and CD105 than those in the smaller microwells. Furthermore, cultures in the 600-μm microwells tended to have higher expression of hypertrophic (Col X) cartilage matrix proteins than those in the smaller microwells. The nonhyaline cartilage proteins produced by cell clusters in the 600-μm wells were expressed at the periphery of the clusters. Thus, hMSC clusters in small microwells tend to undergo hyaline chondrogenic differentiation that is distributed uniformly throughout the cell aggregates. These findings suggest that the hMSC cluster size in the microwells affects the differentiation fate.
In the 600-μm microwells, the cell clusters grew continuously in sphere-shaped clusters until they contacted the walls of the microwells. It was previously reported that tensile stress created at the surface of hMSC clusters is transmitted to the cells 45 ; therefore, this stress may have induced hypertrophic, fibrogenic, and elastic responses that caused increased Col X, Col I, and elastin production in the matrix. A similar phenomenon may also occur for chondrocyte-like cells induced by pellet method. The cell clusters tended to completely fill the spaces within the smaller microwells but not in the 600-μm microwells. Similar to the cells in pellet cultures and large microwell cultures, the cell clusters in the small microwells formed spheres; however, they could not continue to grow because of the constraints imposed by the physical dimensions of the microwells. Therefore, it is likely that hMSC clusters grown in microwells with a diameter equal to or less than 400-μm experience only a small change in surface area with growth, consequently, the lower tensile forces generated by the limited growth may be responsible for the absence of nonhyaline chondrocyte-like phenotypes observed in pellet cultures and 600-μm microwells.
In conclusion, adherent clusters of immortalized hMSCs were cultured in microwells that were coated with a zirconia ceramics. The microfabricated porous zirconia ceramic particles enabled the formation of hMSC clusters that did not attach permanently to the surface. Stimulation of these hMSC clusters with general chondrocyte differentiation medium resulted in a stable induction of the hyaline chondrogenic-like phenotype without the induction of nonhyaline phenotypes that routinely occur in pellet or micro-mass cultures. Our results show that the 3D culture of MSCs in microwells with physical restraints could affect their differentiation fate. In future studies, we plan to harvest the induced chondrocyte-like clusters from the microwell substrata, transplant them in vivo, and regulate their biological function.
Footnotes
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.