
Abstract
Skeletal muscle can be engineered by converting dermal precursors into muscle progenitors and differentiated myocytes. However, the efficiency of muscle development remains relatively low and it is currently unclear if this is due to poor characterization of the myogenic precursors, the protocols used for cell differentiation, or a combination of both. In this study, we characterized myogenic precursors present in murine dermospheres, and evaluated mature myotubes grown in a novel three-dimensional culture system. After 5–7 days of differentiation, we observed isolated, twitching myotubes followed by spontaneous contractions of the entire tissue-engineered muscle construct on an extracellular matrix (ECM). In vitro engineered myofibers expressed canonical muscle markers and exhibited a skeletal (not cardiac) muscle ultrastructure, with numerous striations and the presence of aligned, enlarged mitochondria, intertwined with sarcoplasmic reticula (SR). Engineered myofibers exhibited Na+- and Ca2+-dependent inward currents upon acetylcholine (ACh) stimulation and tetrodotoxin-sensitive spontaneous action potentials. Moreover, ACh, nicotine, and caffeine elicited cytosolic Ca2+ transients; fiber contractions coupled to these Ca2+ transients suggest that Ca2+ entry is activating calcium-induced calcium release from the SR. Blockade by d-tubocurarine of ACh-elicited inward currents and Ca2+ transients suggests nicotinic receptor involvement. Interestingly, after 1 month, engineered muscle constructs showed progressive degradation of the myofibers concomitant with fatty infiltration, paralleling the natural course of muscular degeneration. We conclude that mature myofibers may be differentiated on the ECM from myogenic precursor cells present in murine dermospheres, in an in vitro system that mimics some characteristics found in aging and muscular degeneration.
Introduction
M
Embryonic and induced pluripotent stem (ES/iPS) cell-derived human myogenic cell cultures are already being used successfully as preclinical models of muscular disease.7,8 However, ES/iPS cell-based muscle modeling systems are expensive and time-consuming to develop, and as such, could be complemented by a simpler and more cost-effective adult stem cell-based culture model. Moreover, two-dimensional cellular models do not reflect many aspects of muscular physiology, 9 and it is thus desirable to engineer three-dimensional (3D) tissue constructs that more properly reflect muscle properties in vivo, such as contractility seen on constructs engineered on collagen gels. 10 In this sense, and although a number of 3D tissue-engineered muscle constructs are available, 11 it is currently acknowledged that a suitable cell source (other than satellite cells, which grow poorly in vitro) and improved myogenic differentiation protocols, are urgently needed. 12 It is therefore apparent that an alternative muscle progenitor cell source would be desirable to develop tissue-engineered models, which should have an easier (less invasive) access than muscle. Ideally, and to treat adult onset muscular diseases, the cell source should be available in adults.
While several alternative cell sources are being considered, 13 skin-derived precursor cells (SKPs) meet both these requirements. These dermal precursors may be isolated via minimally invasive procedures.14,15 Furthermore, derivation of skeletal muscle has been achieved from murine SKP cultures, although with relatively low efficiency compared to other myogenic precursor cells.16,17 In this article, by making use of a novel 3D support composed of a mixture of Matrigel-like extracellular matrix (ECM), hyaluronic acid, and netrins, we improve the differentiation of murine dermal precursors to achieve levels of myogenicity similar to those of muscle satellite cells on similar supports.18,19 Future work will pursue derivation of similar engineered muscle constructs from human dermis-derived cells.
Materials and Methods
Animals
Eight-week-old female CD1 mice were used in accordance to the guidelines and approval provided by the Biodonostia Animal Care Committee (San Sebastian, Spain) in accordance with the European Directive 2010/63/EU and the guidelines established by the National Council on Animal Care.
Isolation and proliferation of dermal precursor cells
Precursor cells were isolated from back skin as described. 20 For primary dermosphere expansion, a proliferation medium [Neurobasal™ A (Gibco) supplemented with 2% B27 (Gibco), 1% L-glutamine 200 mM (Sigma-Aldrich), and 1% penicillin/streptomycin] was supplemented every 2 days with a 2% low serum growth supplement (LSGS, Gibco), 40 ng/mL of the epidermal growth factor (EGF; R&D), and 80 ng/mL of the basic fibroblast growth factor (FGF2; R&D).
Preparation of ECM and poly-L-ornithine-coated subtrates
ECM-coated glass coverslips were prepared as described, 21 that is, incubated with a solution of Cultrex® basement membrane extract, hyaluronan, and netrins in phosphate-buffered saline (PBS). To compare ECM-based culture with a previously published protocol, 16 a 0.01% solution of poly-L-ornithine in distilled water (Sigma-Aldrich) was used to coat coverslips, followed by a PBS wash and a drying period of 1 h under laminar flow.
Skeletal muscle induction and contractile myotube video recordings
For muscle induction, primary dermospheres after 7 days of proliferation were gently disaggregated with a 0.25% trypsin-EDTA solution (Sigma-Aldrich) and resuspended in the differentiation medium [the proliferation medium without added growth factors plus 10% fetal bovine serum (FBS; ATCC)], before plating onto coated coverslips at a density of 75,000 cells/cm2. Every 2 days, half of the culture volume was replaced with a fresh medium. At day 5–7 of differentiation, myoblast fusion and contractile activity were observed. Bright field microscopic images of cultures as well as video recordings of twitching myotubes were taken by using a Nikon D90 digital camera coupled to a Nikon Eclipse TS100 microscope.
Skin histology
Murine dorsal skin fragments of approximately 0.5 cm2 were excised, embedded in a Tissue-Tek® O.C.T.™ compound (Sakura), and immediately frozen in isopentane (Merck) cooled in liquid nitrogen. Seven micron transverse sections were processed and stained with hematoxylin–eosin (H&E). 22
Immunofluorescence and microscopy
Cells were washed with PBS (pH 7.2, Ca2+ and Mg2+ free, Gibco) and fixed in 4% paraformaldehyde (PFA; Electron Microscopy Sciences) for 10 min at room temperature (RT). Cells were further washed twice in PBS, and permeabilized/blocked by using 0.3% Triton® X-100 in PBS (PBST) plus 5% normal donkey serum (Sigma-Aldrich) for 1 h at RT. Cells were incubated with the appropriate primary antibody diluted in PBST for 2 h at RT (detailed in Supplementary data; Supplementary Data available online at
Gene expression
Total RNA was extracted from cells by the miRNeasy Mini kit (Qiagen) and converted into complementary DNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to the manufacturer's instructions. Skeletal (gastrocnemius muscle) and cardiac muscle tissues were also used as positive and negative controls for myogenic gene expression analysis, respectively. Real-time quantitative PCR (RT-qPCR) analysis was carried out using TaqMan gene expression assays in the 7900 HT Fast Real-Time PCR System (Applied Biosystems). Each cDNA sample was amplified in triplicates. The cycling conditions were 95°C/10 min followed by 40 cycles at 95°C/15 s, 60°C/1 min in a reaction mixture that contained 1×TaqMan Universal PCR Master Mix and 1×Assay Mix in a final volume of 10 μL. The relative quantity of the gene target was determined by the 2ΔΔCt method. 23
Western blot
Western blots (WB) were performed as described, 24 with minor modifications. Briefly, skeletal (gastrocnemius) and cardiac muscle samples were weighed and homogenized in a TissueLyser mixer-mill disruptor (Qiagen) in 19:1 w/v treatment buffer (0.125 M Tris, 4% SDS, 10% glycerol, 0.1 M EDTA, and 5% β-mercaptoethanol). Similarly, 2×106 cells were lysed in 100 μL of the treatment buffer. About 3×105 of the lysed cells, as well as 5 μg of homogenized samples, were loaded onto SDS-polyacrylamide gels (Bio-Rad).
Transmission electron microscopy
After fixation in 3.5% glutaraldehyde (Electron Microscopy Sciences), cell cultures were washed in 0.1 M PBS (pH 7.4) and treated with 2% osmium tetraoxide (Electron Microscopy Sciences) in 0.1 M PBS (pH 7.4) for 2 h at RT. Samples were rinsed, dehydrated through increasing ethanol solutions, and stained in 2% uranyl acetate (Electron Microscopy Sciences) in 70% ethanol. Dehydrated cell cultures were embedded in araldite (Fluka). Semithin sections (1.5 μm thick) were cut with a diamond knife and stained with a 1% Toluidine blue solution (Sigma-Aldrich), re-embedded for ultrathin (70 nm-thick) sectioning, and examined under a Tecnai-Spirit Transmission Electron Microscope coupled to a Morada TEM CCD camera (Soft Imaging System).
Electrophysiological recordings, data acquisition, and analysis
Electrophysiological properties of twitching myotubes were investigated after 10–14 days in culture using whole-cell patch-clamp recording techniques. 25 Acetylcholine (ACh)-induced currents, action potentials, and membrane potential changes were recorded using the perforated patch configuration 26 under either current or voltage clamp mode, respectively. Myotube-containing, ECM-coated coverslips were placed on a recording experimental chamber mounted on the stage of a Nikon Eclipse T2000 inverted microscope. A perforated patch was obtained using borosilicate glass pipettes (Kimble Chase) containing 50–100 μg/mL amphotericin B in DMSO (both from Sigma-Aldrich) as a permeabilizing agent, 27 and a pipette-filling intracellular solution (freshly prepared every 2 h) containing (in mM) 135 KCl, 10 NaCl, 2 MgCl2, 10 HEPES, and 5 EGTA (pH 7.3 with KOH) (all from Sigma-Aldrich). To facilitate sealing, the pipette was first dipped in a beaker containing the internal solution, and then back-filled with the same solution containing amphotericin B. Recordings started when access resistance decreased below 15 MΩ, which usually occurred within 10 min of sealing. Voltage and current recordings were made with fire-polished electrodes (resistance 2–5 MΩ when filled with the intracellular solution) mounted on the headstage of an EPC-10 patch-clamp amplifier (HEKA Electronic), allowing cancellation of capacitative transients and compensation of series resistance. Data were acquired with a sample frequency of 5 kHz. Recordings with leak currents >100 pA or series resistance >20 MΩ were discarded. During recording, myotubes were locally, rapidly, and continuously superfused with a Tyrode's solution containing (in mM) 2 CaCl2, 137 NaCl, 1 MgCl2, 10 glucose, 5.3 KCl, and 10 HEPES/ NaOH (pH 7.3; all from Sigma-Aldrich). The different drugs (ACh; tetrodotoxin, TTX; d-tubocurarine, dTC) were dissolved in the extracellular solutions, which were rapidly exchanged using electronically driven miniature solenoid valves coupled to a multibarrel concentration-clamp device, the common outlet of which was placed within 100 μm of the cell to be patched. The flow rate was 1 mL/min and was regulated by gravity. Data acquisition was performed using PULSE programs (HEKA Elektronic) while data analysis was performed using GraphPad Prism software (version 5.01 for Windows). All experiments were performed at room temperature (22°C–24°C). At least three independent experiments were performed to determine the significance of statistical results.
Measurements of cytosolic Ca2+ concentration ([Ca2+]c) changes, data acquisition and analysis
The setup for fluorescence recordings was composed of a Leica DMI 4000 B inverted light microscope (Leica Microsystems) equipped with an oil immersion objective (Leica 40×Plan Apo; numerical aperture 1.25). Before assessment, cells were incubated for 1 h at 37°C in the differentiation medium containing the calcium probe Fura-2 AM (10 μM; Invitrogen). After Fura-2 loading, coverslips were mounted in a chamber, and washed/covered with the Tyrode's solution (as above). Cells were continuously superfused by means of a five-way superfusion system at 1 mL/min with a common outlet 0.28-mm tube driven by electrically controlled valves with the Tyrode's solution with/without drugs (ACh; nicotine; histamine; caffeine; nifedipine; d-tubocurarine, dTC; all from Sigma-Aldrich). Fura-2 AM was excited alternatively at 340±10 and 387±10 nm using a Leica Küber CODIX xenon lamp and emitted fluorescence was collected through a 540±20 nm emission filter and measured with an intensified charge coupled device camera (Hamamatsu camera controller C10600 orca R 2 ). Fluorescence images were generated at 1-s intervals. Images were digitally stored and analyzed using LAS AF software (Leica). Data analysis was carried out on a personal computer, and data obtained from LAS AF software were exported to Microsoft Excel tables. Graphs and mathematical analyses were performed using the Graphpad Prism software (version 5.01). Areas or peak heights were calculated by integrating the calcium transient over time during the stimulus duration by means of Origin Pro 8 SR2 software (version 8.0891; OriginLab Corporation). Areas were worked out by the integration of the input data set by using the trapezoidal rule. Results are expressed as mean±SEM.
Adipogenic assays
Adipocytes were assessed by Oil Red O staining. 28 Cultures were fixed twice in 10% formalin (Sigma-Aldrich) for 10 min and 1 h at RT, respectively. Fixed cultures were rinsed sequentially with distilled water and 60% isopropanol (Sigma-Aldrich) for 5 min at RT. Once completely dry, cells were stained with the Oil Red O working solution [60% stock solution (0.35% Oil Red O (Sigma-Aldrich) in isopropanol) and 40% distilled water] for 10 min at RT. After four consecutive washes with distilled water, images were acquired. Water was then removed and Oil Red O eluted by adding 100% isopropanol for 10 min under gentle shaking. Optical absorbance (OD) at 500 nm was measured using 100% isopropanol as a blank. Three individual experiments were performed and results were expressed as fold change respect to the basal condition (24 h after culture, absence of adipocytes). The total concentration of triglycerides was determined by a coupled enzyme assay (Adipogenesis Assay Kit; Sigma-Aldrich), which results in a colorimetric (570 nm) product proportional to the triglycerides present in each sample.
Assessment of statistical significance
Statistics carried out using GraphPad Prism software. A one-way analysis of variance (ANOVA) with subsequent pairwise multiple comparison procedures (Bonferroni's test) was used to assess the statistical significance of the results from immunocytochemistry, RT-qPCR, and adipogenesis experiments. Unless otherwise stated, statistical analyses of patch-clamp results were carried out with the one-way ANOVA test followed by Tukey's post hoc analyses. Single or multiple cytosolic Ca2+ concentration ([Ca2+]c) changes were carried out with a one-tailed paired t-test for a confidence interval of 95% or the one-way ANOVA test followed by Dunnett's post hoc analyses, respectively. The signs *, **, or *** represent a statistical significance of p<0.05, p<0.01, or p<0.001, respectively.
Results
Spontaneously twitching myotubes engineered from dermis-derived precursors
Once adult dermal precursors that present myogenic potential (both in vitro and in vivo) as previously described16,17 were obtained, two major questions remain unanswered: (1) what is the source of myogenic precursors within murine skin, and (2) can we improve the relatively minor differentiation potential shown in vitro by these precursors? To shed light on both issues, we set up an improved isolation, proliferation, and differentiation protocol for dermis-derived precursors as depicted in Figure 1A. Of note, primary cell preparations of disaggregated murine skin include fragments of Panniculus carnosus (PC) muscle (Fig. 1B–D). After 7 days of proliferation, dermis-derived precursors generated spheres, while muscle fragments were no longer visible in culture (Fig. 1E). Dermal spheres, enriched in precursor cells, were then seeded onto ECM-coated coverslips, and multinucleated myotubes were observed after 5–7 days in the presence of serum (Fig. 1F, G). Interestingly, the engineered myotubes twitched spontaneously, at times in relative isolation (Supplementary Fig. S1; Supplementary Data are available online at
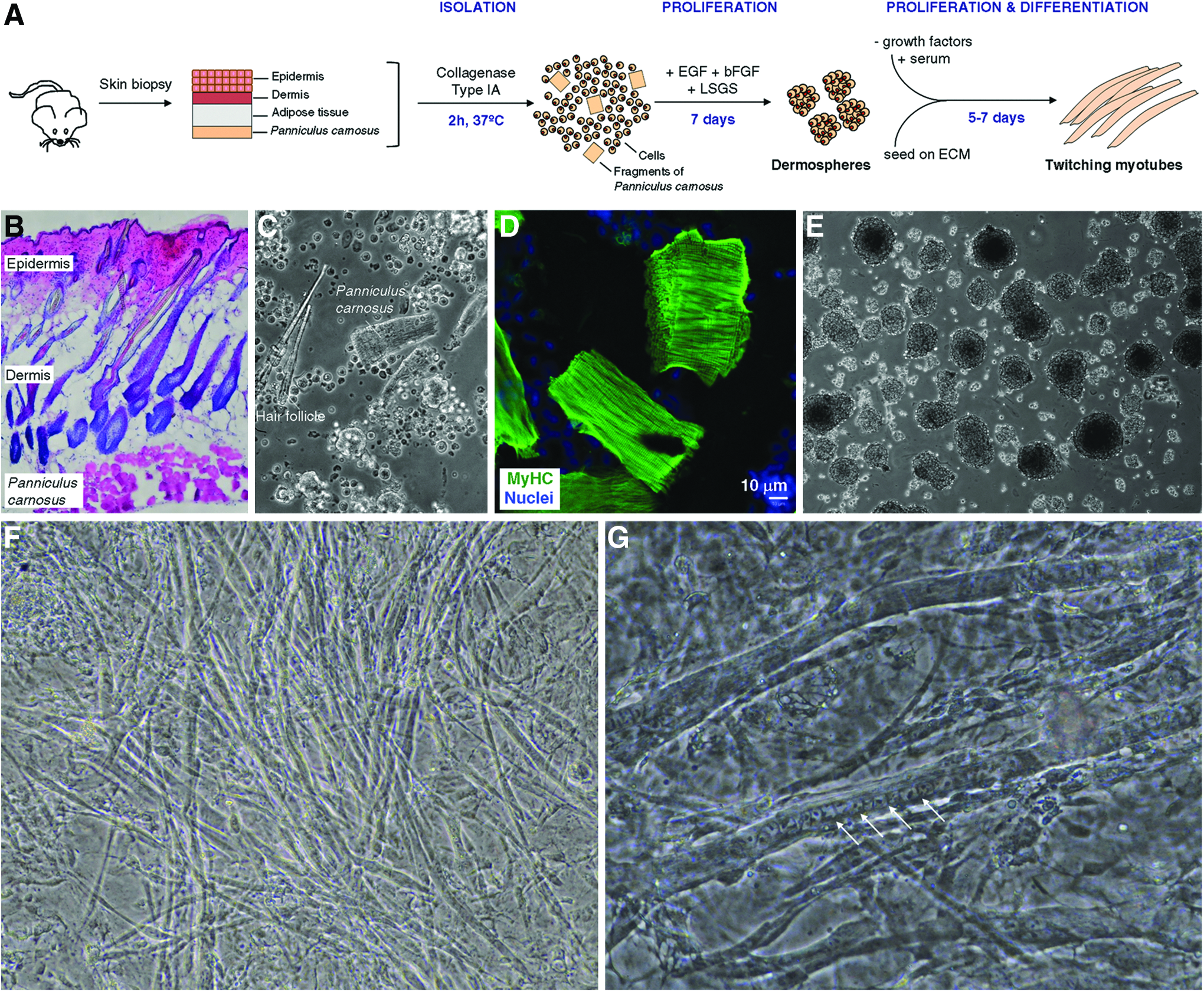
Twitching myotubes engineered from dermal precursor cells.
To characterize the appearance of myogenic markers in dermal proliferation culture, expression of Pax7, MyoD1, Myogenin (Myog), and MyHC were analyzed at day 0 and 7 of sphere culture by Western blot (Fig. 2). As expected (since muscle fragments were present straight after tissue disaggregation), myogenic markers Pax7, MyoD1, Myog, and MyHC (all fibers) were readily detected at day 0. In contrast, embryonic MyHC, a marker of myogenic precursors and nascent myotubes, was absent. Interestingly, expression of all these markers diminished (to levels barely detectable by Western blot) by day 7 when dermospheres were already fully formed. The only exception being embryonic MyHC that was clearly detected at this stage (Fig. 2A, B). These data thus confirmed that muscle tissue remnants appeared in dermal cultures at day 0, but not at day 7, and suggested the appearance/enrichment of a myogenic precursor during proliferation culture. To further validate protein expression data, messenger RNA levels for the genes Pax7, MyoD1, Myog, MyH2 (adult myosin), and MyH3 (embryonic myosin) were analyzed at day 7 relative to day 0 by real-time quantitative PCR (RT-qPCR, Fig. 2C). The results confirmed a reduction of myogenic gene mRNA levels as culture progressed, with the exception of embryonic MyHC that was stable or augmented with culture progression. To discriminate the proportion of sphere cells adopting myogenic commitment, expression of MyoD1, Myog, MyHC, TnT, and Serca1 was analyzed by immunofluorescence in day 7 dermospheres (Fig. 2D). Interestingly, a discrete subpopulation of cells was found to express myogenic markers, while the bulk of the precursor cells in the sphere remained negative at day 7. Taken together, these results suggest that twitching myotubes derive from a discrete population of myogenic precursors present in dermal spheres at day 7 of proliferation.
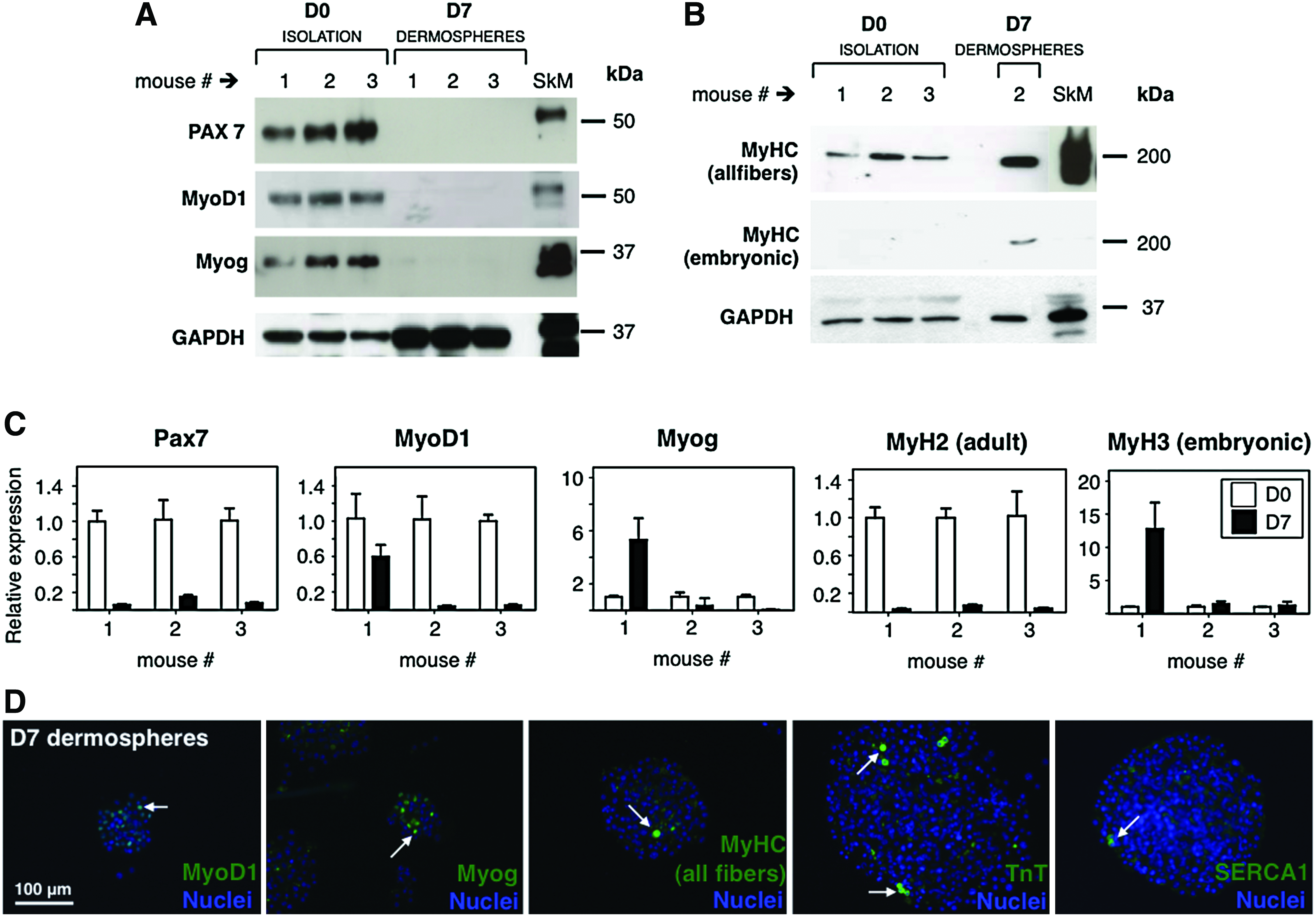
Presence of myogenic markers in dermal spheres at the proliferation phase.
Myotubes present structural characteristics of bona fide skeletal muscle
To further characterize in vitro engineered myotubes, the expression and subcellular localization of myogenin and MyHC were analyzed by immunofluorescence (Fig. 3). As expected, multinucleated myotubes that express nuclear Myog were readily detected (Fig. 3A, B). Myotubes showed the characteristic striated pattern of MyHC, as predicted for sarcomeric proteins (Fig. 3C, D). Since SKPs are neural crest derived, and similar tissue-resident precursors have been shown to differentiate into cardiomyocites, 29 we wondered if the observed striated phenotype might correspond to the cardiac, not skeletal muscle. For this reason, we analyzed expression of skeletal or cardiac muscle-specific ryanodine receptors by RT-qPCR (Fig. 3E). Skeletal muscle-specific RyR1 mRNA was readily detected, while cardiac muscle-specific RyR2 remained undetectable as compared to control samples of both striated muscles. Cardiac-specific TNNI-3 was also absent from engineered myotube cultures, as assessed by Western blot (Fig. 3F).
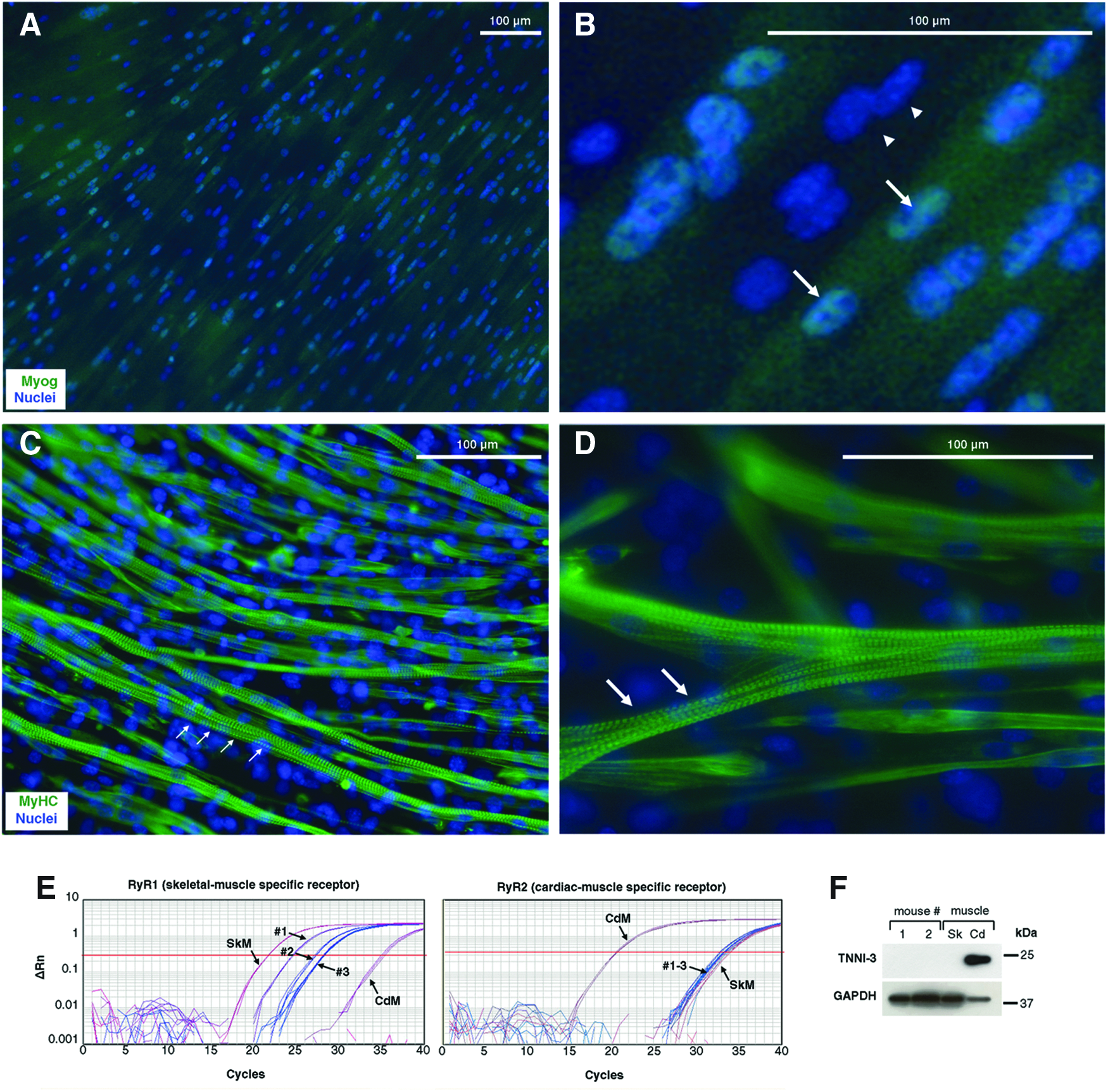
Engineered muscle is composed of skeletal (not cardiac) myotubes.
Ultrastructural analyses were then performed by transmission electron microscopy (TEM) (Fig. 4). Semithin (1.5-μm) sections of resin-embedded tissue constructs showed cells of either spherical or elongated morphology (Fig. 4A). Spherical cells typically were found with irregularly shaped multiple nuclei that were centrally positioned and abundant organelles scattered through the cytoplasm, with frequent detection of small vacuoles. Some of the spherical cells had a smaller size, irregular cell shape, scarce cytoplasm, and very dense nuclei (not shown). In contrast, elongated cells (Fig. 4B) were tubular as clearly visualized on transversal sections (Fig. 4C). Elongated (tubular) cells presented no expansions in contrast to spherical cells. Tubular cells showed a number of centrally positioned nuclei, each having several nucleoli. Interestingly, some elongated cells showed striation of dense and light bands (Fig. 4B). By TEM, the classical actomyosin structures characteristic of striated muscle were clearly visible (A- and I-bands; H zone and Z-line in the sarcomeres; Fig. 4D, E). Repeated measurements showed that sarcomeres were 1.5–3.3 μm long, A-bands 1–2 μm, and I-bands 0.5–1.3 μm. Overall, actomyosin fibers were 0.3–0.6 μm thick. Prolonged mitochondria running parallel to the fibers were often visible; some of them were branched, with abundant crests (Fig. 4D, E), and were also aligned to smooth endoplasmic reticulum (SER). Besides, SER cisternae surrounded actin and myosin bundles in transversal sections (Fig. 4F), as well as transversally oriented mitochondria. Golgi bodies scattered throughout the cytoplasm were also found. Unstriated fusiform cells, which presented abundant dilated rough endoplasmic reticulum (RER) cisternae, were identified as possible fibroblasts. In contrast, striated muscular cells presented scarce, generally undilated RER. Muscle fibers showed no branching and occasionally adherens junctions were seen between adjacent cells (Fig. 4G). Abundant caveolae were seen on the cell surface in transversal sections (Fig. 4H). These results unequivocally demonstrate that skeletal (not cardiac) muscle-like myotubes are being engineered in vitro that fully recapitulate ultrastructural characteristics of bona fide skeletal muscle.
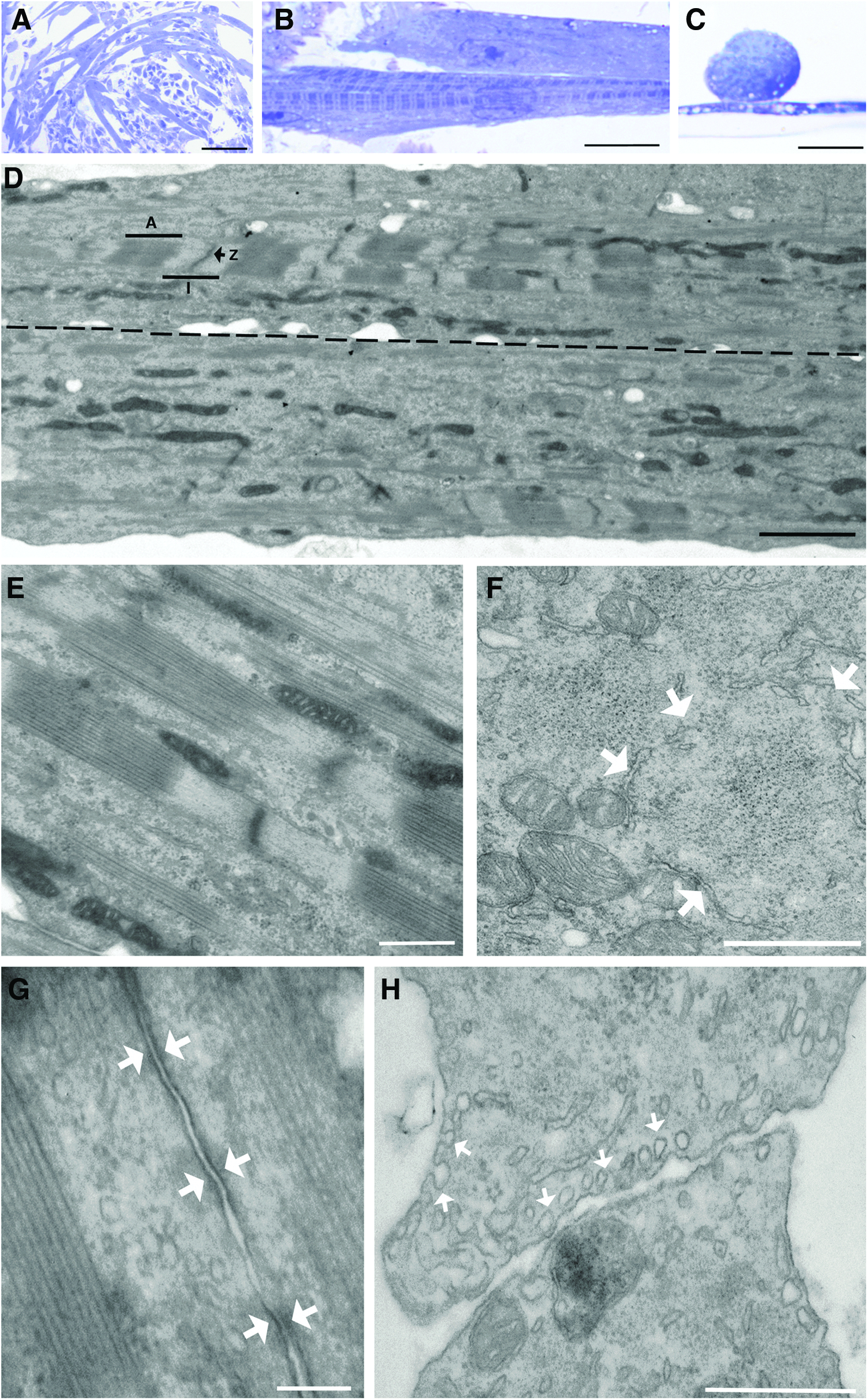
Ultrastructural characterization of myotubes.
Electrophysiological characterization of in vitro engineered myotubes
Once characterized by immunocytochemistry and specific biomarkers, we pursued functional studies to determine the nature of the cells under study. Electrophysiological experiments were performed by patching dermis-derived myotubes and challenging them with 100 μM ACh pulses of 250 ms duration (Fig. 5). ACh evoked a 2 nA inward current when membrane potential was clamped at −80 mV. After rapid activation kinetics, slow inactivation developed lasting for about 5 s (Fig. 5A). ACh current charge (QACh) were carried by Na+ and Ca2+ ions, so its substitution by the same mOsm quantity of N-metyl-D-glucamine resulted in a strong reduction in QACh and IACh peak (Fig. 5B). In current clamp configuration, a twitching myotube fired spontaneous action potentials that were abolished in the presence of the Na+ channel blocker TTX (Fig. 5C). To further characterize the nature of the receptor implicated in this signal, we tested the neuromuscular blocker d-tubocurarine in our preparation. At 10 μM, this drug reversibly blocked IACh and QACh by 90% approximately (Fig. 5D).
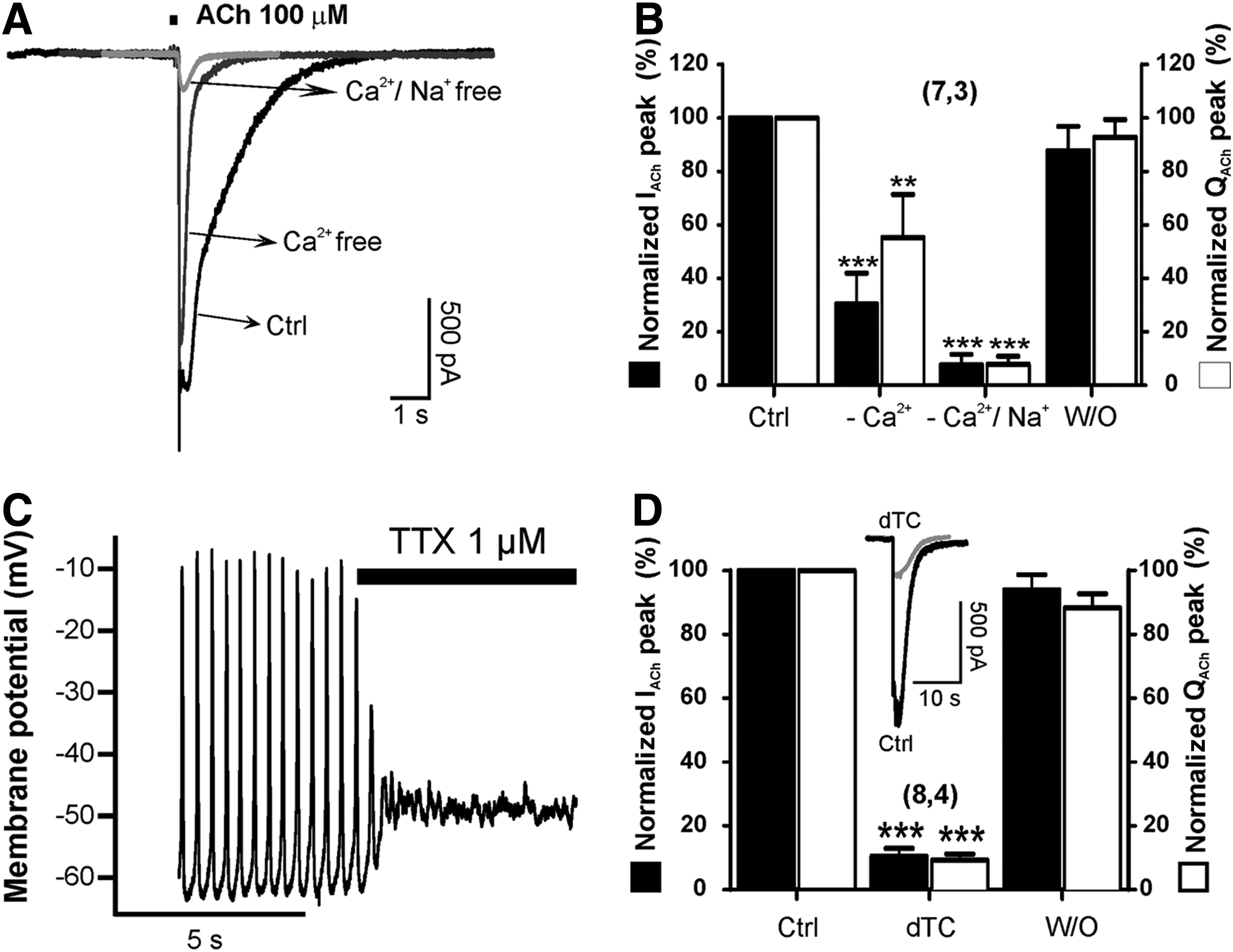
Acetylcholine-elicited currents (IACh) and electrical excitability of differentiated beating myotubes.
Muscle contraction is a Ca2+-dependent process, which in turn depends on cell depolarization and Ca2+-induced Ca2+ release (CICR) from the sarcoplasmic reticulum. 30 So, we wanted to look at cytosolic Ca2+ signaling ([Ca2+]c) mediated by ACh receptor stimulation (Fig. 6). Cells were incubated for 1 h with Fura-2AM. Under fluorescence recording, cells stimulated with ACh increased their [Ca2+]c as shown in Figure 6A and Supplementary Movie SM4. Each photogram is taken at different time points after application of the 100 μM ACh pulse. These cells were tested against other stimuli, namely, nicotine, caffeine, and histamine. Twitching cells responded to 30 μM nicotine, 20 mM caffeine, but not to 100 μM histamine (Fig. 6B, C and Supplementary Table S1). However, another cell population with different morphology (not myotubes), also present in dermis-derived muscle cultures, had the opposite response pattern increasing their [Ca2+]c in response to histamine, but not to caffeine or nicotine (Fig. 6D, E and Supplementary Table S1). At this time point, the question of whether the observed [Ca2+]c increase was a dihydropirydine-dependent effect arose. So, we measured ACh-mediated [Ca2+]c in the presence of 3 μM nifedipine and congruently found that calcium peak amplitudes were unaffected by nifedipine incubation (Fig. 6F and Supplementary Table S2). In contrast, 10 μM d-tubocurarine significantly blocked the ACh-induced [Ca2+]c increase (Fig. 6G and Supplementary Table S3). These results demonstrated that beating myotubes exhibited characteristics compatible with a skeletal muscle fiber nature, namely, (1) d-tubocurarine-sensitive and Na+- dependent inward currents generated by the muscle end plate physiological neurotransmitter ACh, (2) ACh-elicited cytosolic Ca2+ transients that are also blocked by d-tubocurarine, and (3) caffeine-elicited [Ca2+]c transients compatible with sarcoplasmic reticula Ca2+ release mediated by RyR1 receptors.
Myogenicity of ECM-based cultures as compared to previous differentiation protocols
Although several reports have addressed myogenicity of dermal cells, quantitative data are often lacking. Due to the impressive, widespread contractility observed on ECM-based cultures, we were interested in performing a quantitative comparison with previously published differentiation protocols. To this end, we chose the protocol based on the use of 5-azacytidine as an inducer of myogenic differentiation. 16 Parallel differentiation cultures were set up and direct comparison was performed by RT-qPCR and immunofluorescence analyses (at day 1 vs. day 12 of differentiation, Figure 7). As expected, mRNAs for Pax7, MyoD1, Myog, MyH2, and MyH3 were clearly overexpressed in ECM-based cultures as compared to 5-azacytidine-based cultures (Fig. 7A). Immunofluorescence analyses demonstrated a major increase in MyHC(+) myotubes when the ECM was used as a substrate, although there was no significant difference on Myog levels (Fig. 7B–D). These results pointed to a faster generation of dermis-derived myotubes on the ECM as compared to 5-azacytidine-based cultures.
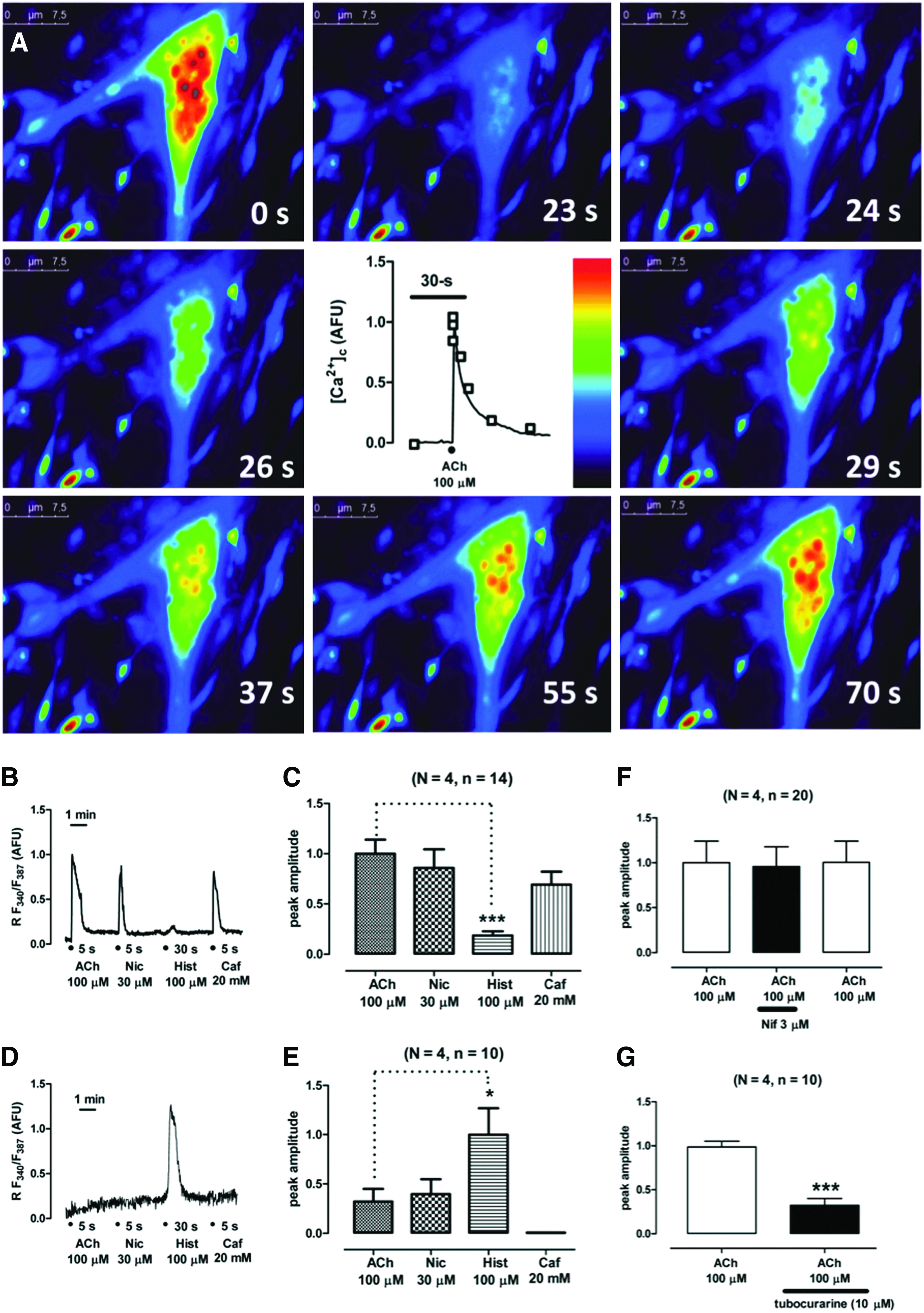
Characterization of cytosolic Ca2+ transients elicited by challenging myotubes with various stimuli. Cells were loaded with the calcium probe Fura-2 AM and were subsequently perifused with various saline solutions containing different stimulating compounds.
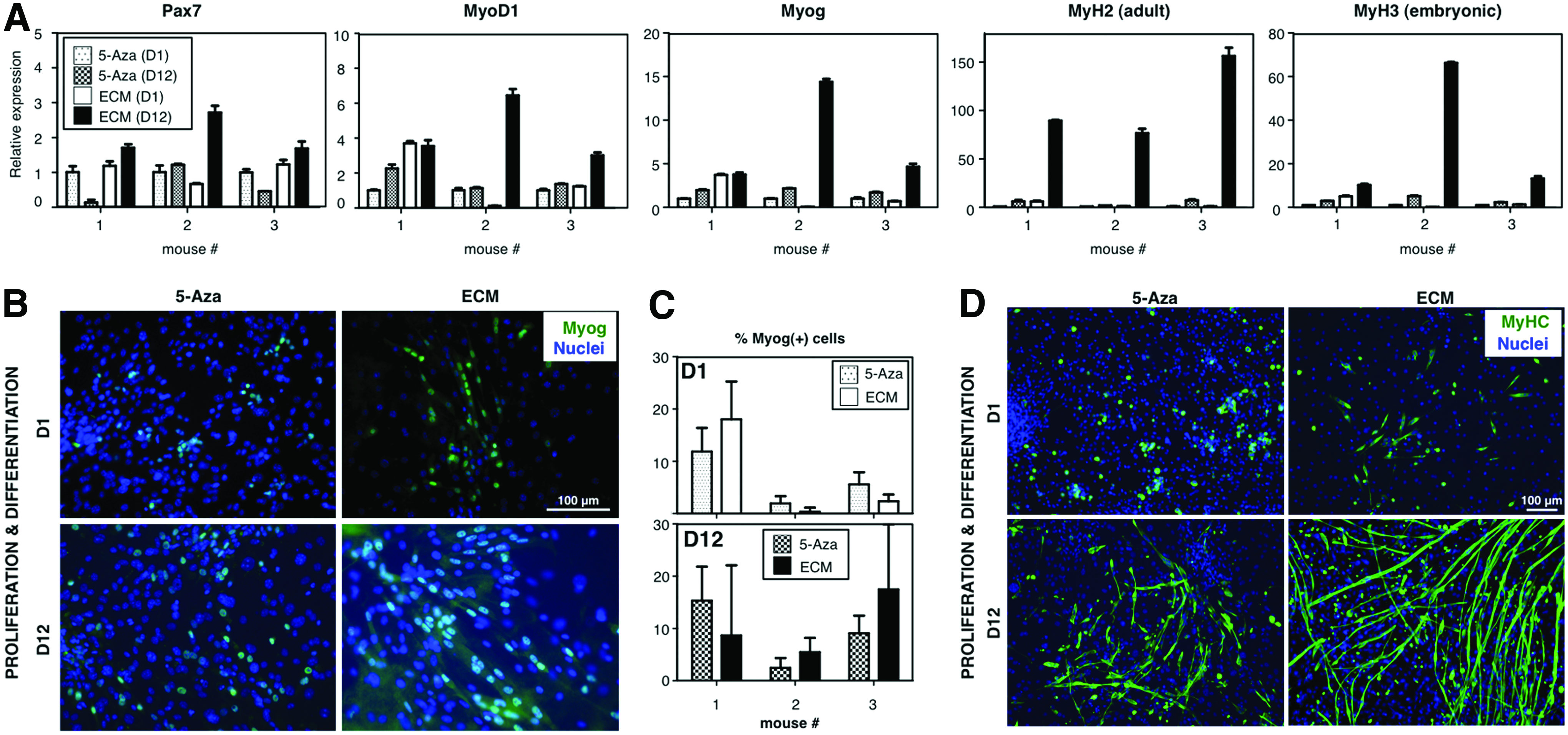
ECM-based differentiation cultures show enhanced myogenicity. In parallel differentiation cultures run for 12 days, ECM-based cultures were compared to 5-azacytidine-based counterparts.16
Long-term evolution of ECM-based cultures
While satellite cell-derived myotubes grown on Matrigel show great resilience in vitro, 18 we observed that ECM-based, dermis-derived cultures, which were derived from complex cellular mixtures, degraded over time (Fig. 8). To characterize this phenomenon, we investigated expression of myogenic mRNAs Myog and MyH2 over a 28-day period of differentiation. Interestingly, myogenin peaked at day 7, while adult myosin expression levels reached their highest at day 14, both of them decreasing sharply afterward (Fig. 8A). Of note, muscle degeneration is concomitant with fatty infiltration in vivo, and a PDGFRα+ precursor cell population, which resides in muscle, but is distinct from satellite cells, seems to be responsible for this phenomenon. 31 For these reasons, we checked PPARγ, AdipoQ, and FABP4 adipogenic gene expression during the same time frame. Strikingly, adipogenic genes peaked at day 14 to 21, concomitant with loss of myogenic gene expression (Fig. 8A). Accordingly, adipocytes were visible by Oil Red O staining by day 7, and abundantly so after day 21 (Fig. 8B). Adipocytes matured in culture over time, with increased fusion of cytoplasmic lipid vesicles. Moreover, quantitative analyses of Oil Red O and trygliceride content showed a sustained increase in culture over time (Fig. 8C, D). In summary, these results are consistent with degradation of the muscle constructs in vitro (Fig. 8E) that is coincident in time with the appearance of fat tissue.
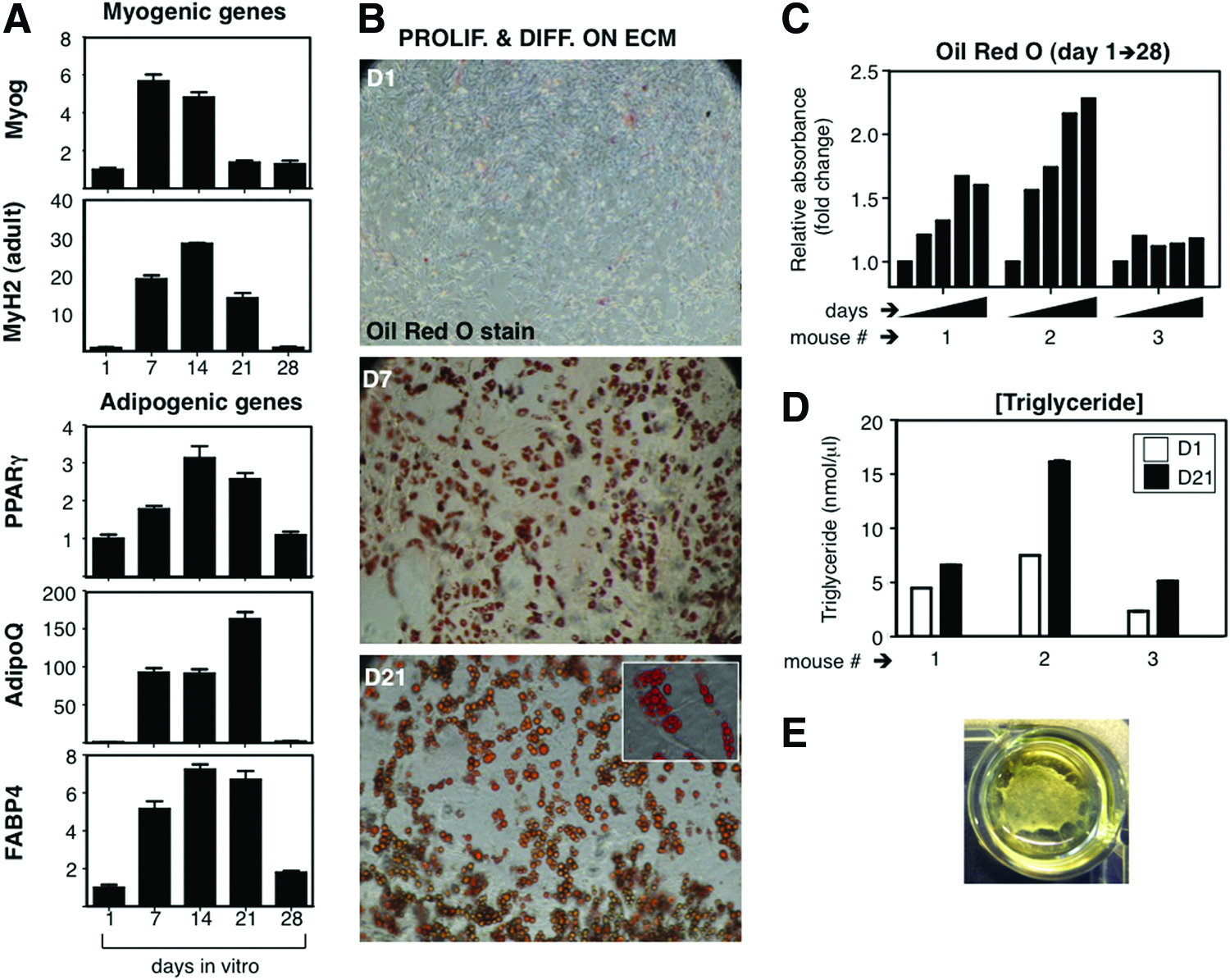
Long-term progression of myotube cultures shows fatty infiltration.
Discussion
In this study, we improved previously published protocols to engineer the skeletal muscle from dermal precursor cells.16,17 Underlying this improvement in muscle quantity and quality (at least in vitro), the use of a Matrigel-like platform might have been critical. 32 Muscle organoids have been previously derived from primary neonatal rodent myoblasts suspended in a 1:6 solution of Matrigel: Collagen type I. 33 However, several issues remain unresolved.
For a start, the nature and origin of the discrete population of myogenic precursor cells present in primary dermospheres is still unclear. We have shown that Panniculus carnosus (PC), a vestigial muscle in mammals that is often neglected by the literature, 17 is present in murine dermal cell preparations. PC muscle derives from Pax7+ cells, as demonstrated when these progenitors were genetically traced both at the E9.5 (multipotent stem cells of the dermomyotome) and E11.5 (cells restricted to the myogenic lineage) developmental stages. 34 On the other hand, skeletal muscle regeneration consists of the fusion of myoblasts for de novo myotube formation, generated not only by satellite cells, but also by a number of other cell types such as bone marrow-derived mesenchymal stromal cells, muscle side population cells, and pericytes. 35 In murine models, PC has been shown to possess a higher regenerative activity than most skeletal muscles, with significantly smaller fiber diameters, increased heterogeneity of the fiber size, and a high percentage of centrally nucleated myofibers in the absence of focal injury. Further, PC myofibers present the highest rate of bone marrow-derived cell incorporation. 36 An enticing possibility that requires further investigation is that the myogenic precursors in PC originate in the bone marrow, and that bone marrow-derived cells underlie the phenomena observed when dermospheres were put under myogenic stimulation.
A second question that remains unanswered is the origin of the fatty infiltration that we observed. Adipogenic differentiation is detected in isolated myofibers, suggesting that satellite cells or other progenitors that reside in that same niche contribute to fat formation. 37 Furthermore, the existence of fibro/adipogenic progenitors (FAP) in vivo, which are perivascular localized and distinct from satellite cells, has been postulated. 31 It is therefore plausible that such FAPs might also be present in other interstitial tissues such as the dermis, although PPAR and FABP detection may not directly reflect the presence of FAPs, but truly relate to fatty infiltration. Since an intact niche is key to the maintenance of the satellite stem cell pool, 38 an attractive hypothesis would be that ECM degradation provoked by myotube maturation could alter the balance of the transition from myogenesis to adipogenesis. 39 However, experimental evidence for this proposal is currently lacking. In any case, the engineered muscle system presented here represents a major improvement over modeling systems where muscle ageing is mimicked by making use of high passage myoblast cell lines. 40
The generalized contractile phenotype we observed was highly reminiscent of that described for chicken myogenic cultures grown on Matrigel, in the absence of innervation. 18 In contrast, human myotubes in monolayer culture present sparse spontaneous contractions that may be improved by motor neuron innervation. 41 For this reason, cocultures of spinal cord explants with myofibers have long been performed to achieve functional innervation and contractile behavior. 42 More recently, neuromuscular junctions have been engineered in vitro from human ESC-derived cocultures.43,44 Given the fact that the very same ECM/dermal precursor combination we used in this article has been shown to efficiently generate neural progeny,20,21 the following question arose: are in vitro engineered myotubes innervated by motor neurons present in dermis-derived cultures? The most plausible answer is no, since no neurons were present in the original dermal cell starting culture and myotube contractions are visible very early, only a few days after differentiation starts. On the other hand, we detected nicotinic ACh receptor (nAChR) clusters on myotubes (Supplementary Fig. S2), reminiscent of those seen in neuromuscular junctions; in fact, stimulation of these receptors with ACh generates inward ACh currents and [Ca2+]c signals very similar to those found in control skeletal muscle. 30 However, clusters of nAChR are present in extrajunctional areas of tissue-cultured embryonic muscle. 45 Furthermore, prepatterned nAChR clusters are known to be required for twitching of developing myotubes, through autocrine activation.46,47 Alternatively, non-neuronal cells present in the dermis could be the source of ACh in these cultures. 48
Ideally, the engineered muscle of human (not murine) origin should be employed to test novel therapeutics. It might follow that, since SKPs are also present in human beings, engineered human muscle could also be produced in a similar fashion. Our data suggest that myogenic precursors are present early after isolation, possibly related with the existence of muscle fragments in the original disaggregated cell mixture. These myogenic precursors could thus originate in the PC, a vestigial organ in humans. Although further research is needed to ascertain this point, if muscle-derived progenitors are eventually required for this process, then human skin should be obtained from the limited areas of the body that present this vestigial muscle. 49 First of all, mouse lineage-tracing experiments should be performed that shed further light on the cell lineage originating dermis-derived muscle. Secondly, if PC-derived myogenic precursors are traced in the murine models, a confirmation of mouse data should be obtained by using human PC-derived cells. This might prove difficult because of limited PC availability in the human body and the necessity to perform full-thickness skin biopsy that reaches up to the fascia. A promising approach might be isolation of stem cells from human cadavers as recently reported. 50 Independently of the potential difficulties in extending this model to human cells, we present a robust model, genetically unmodified, that may be of use to model a number of muscular diseases using genetically modified mice as a source of skin cells. Furthermore, the unique ability to model fatty infiltration as seen after natural muscle atrophy should be of great use to test novel pharmacological approaches to age-related fragility.
Footnotes
Acknowledgments
We thank Charles Lawrie for critical reading of the manuscript. We thank investigators for monoclonal antibodies A4.1025 and F1.652 (H.M. Blau), F5D (W.E. Wright), Pax7 (A. Kawakami), RV-C2 and TI1 (S. Schiaffino), which were obtained from the Developmental Studies Hybridoma Bank (developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242). Research in A.I.'s laboratory and a postdoctoral contract for P.G.-P. was supported by grants from FIS and INNPACTO programs (PI10/02871 and IPT-300000-2010-17, provided by Ministerio de Ciencia e Innovación) and Diputación Foral de Gipuzkoa (OF 53/2011 and OF 98/2012). A.I. was supported by the “Programa I3SNS” (CES09/015) from Instituto de Salud Carlos III (ISCIII) and by Osakidetza-Servicio Vasco de Salud (Spain). A.L.M. received research support by the Association Francaise contre les Myopathies (Ref. 12642), the Spanish Ministry of Health (FIS PS09-00660), the Ilundain Foundation, Isabel Gemio Foundation, Diputacion Foral de Gipuzkoa (DFG09/001), and SAIOTEK (SAIO12-PE12BN008). A.A. was supported by ISCIII (CA00/01506; Ministerio de Economia y Competitividad) and Instituto Biodonostia. M.G. was supported by FIS (PS09-00660) and by Ilundain foundation. M.M., J.F.P., J.C.F-M., and A.G.G. were supported by (1) SAF 2010-21795, Ministerio de Economía y Competitividad (Spain); (2) RENEVAS-RETICS-RD06/0026, ISCIII (Spain); and (3) CABICYC, UAM/Bioibérica (Spain). We thank the continued support of Fundación Teófilo Hernando of Madrid (Spain).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.