
Abstract
The vascularization of tissue-engineered constructs is yet an unsolved problem. Here, recent work on the decellularization of whole organs has opened new perspectives on tissue engineering. However, existing decellularization protocols last several days and derived biomatrices have only been reseeded with cells from the same tissue origin or stem cells differentiating into these types of tissue. Within the present work, we demonstrate a novel standardized, time-efficient, and reproducible protocol for the decellularization of solid tissues to derive a ready to use biomatrix within only 5 h. Furthermore, we prove that biomatrices are usable as potential scaffolds for tissue engineering of vascularized tissues, even beyond tissue and maybe even species barriers. To prove this, we seeded human primary osteoblasts into a rat kidney bioscaffold. Here, seeded cells spread homogeneously within the matrix and proliferate under dynamic culture conditions. The cells do not only maintain their original phenotype within the matrix, they also show a strong metabolic activity and remodel the biomatrix toward a bone-like extracellular matrix. Thus, the decellularization technique has the ability to become a platform technology for tissue engineering. It potentially offers a universally applicable and easily producible scaffold that addresses the yet unsolved problem of vascularization.
Introduction
T
In 2004, Mertsching and her group presented a promising idea. They decellularized intestinal segments that were harvested together with their vascular supply to generate a scaffold for tissue engineering with an inherent vascular network. They could provide proof-of-principle with successful decolonization of the vascular network with endothelial cells and survival of seeded stromal cells under dynamic culture conditions. 3 Within the last 3 years, the decellularization method was translated to solid organs with great success. 4 After decellularization and reseeding with cardiomyocytes, a perfused, pumping rat heart could be generated in vitro. 5 Uygun et al. reported the generation and successful transplantation of a recellularized liver graft in a rat model. 6 Furthermore, decellularized lungs could be successfully reimplanted after recolonization and provided gas exchange in vivo in a rat model.7,8
The extracellular matrix (ECM) provides an important function in native tissue. It is part of the microenvironment—the so-called cell niche—which regulates the differentiation and proliferation of the cells that reside in it. 9 This function is mediated by integrin and cytokine signals within the matrix. Recent work revealed that this function of the ECM is partially preserved during the decellularization process. This induces pluripotent cells seeded on decellularized scaffolds to differentiate into tissue-specific cell types.10,11 In the future, this cell differentiation enabling activity of the ECM may even allow tissue engineering of complex tissues, which are composed of different cell types. On the other hand, the composition of the ECM is dynamic and constantly changing in response to the metabolic activity of the resident cells, which are permanently modulating their own niche. 12
This reported plasticity of ECM composition led us to the hypothesis that vascularized bioscaffolds can be utilized as universal matrices for tissue engineering, regardless of tissue origin. According to our hypothesis, differentiated cells seeded on the donor matrix will adapt the scaffold to their niche as long as applied differentiation stimuli are sufficient to maintain the differentiation of the cells. To address this hypothesis, we first set up a time-efficient and effective decellularization protocol to prepare decellularized rat kidneys. Subsequently, we seeded human osteoblasts into the rat kidney as a model for a species- and tissue-dissimilar cell type. To investigate our hypothesis, the constructs were monitored over a period of 2 weeks to prove survival, proliferation, phenotypic and metabolic activity of the cells.
Materials and Methods
The culture medium and supplements were purchased from Biochrom AG and all other chemicals were obtained from Sigma, Germany, unless stated otherwise. Human umbilical vein endothelial cells (HUVEC) and the endothelial cell medium were purchased from Promocell.
Ethical statement
All used osteoblasts were isolated from patients undergoing total hip replacement in the Department of Orthopedics, Klinikum rechts der Isar, Technical University Munich. The study was approved by the ethics committee of the faculty of medicine of Technical University Munich, Germany. The patients provided their written consent before surgery.
Decellularization of rat kidneys
Rat kidneys were obtained from cadavers of adult Sprague-Dawley rats weighting approximately 500 g. Within 30 min after the rats were euthanized, the kidneys were harvested without previously administering anticoagulation medication to the animals. After harvesting, the kidneys were frozen at −80°C in phosphate-buffered saline (PBS). They were kept in this state for 4 to 12 weeks until they were used for experiments. Before decellularization, the kidney was thawed and the surrounding soft tissue was carefully removed. A cannula was inserted into the renal artery and fixed with nonresorbable surgical suture (Ethicon). The organ was then connected to an arthroscopic pressure-controlled roller-pump Arthrex AR-6475 using the sterile standard arthroscopy tube system (Arthrex).
The decellularization procedure of the organ was performed with a pressure of 100 mmHg at room temperature. The kidneys were perfused with distilled water for 10 min. This step was followed by perfusing the organ with the sodium dodecyl sulfate (SDS) solution. For the first part of the study with experiments to establish a time-efficient decellularization protocol, SDS concentrations of 0.25%, 0.5%, 0.66%, and 1% were used, combined with a perfusion time of 0.5, 1, 2, and 4 h. For the second part of the study—the reseeding experiments—the concentration of SDS was always 0.66% and the perfusion time was 1 h. After the first 30 min of perfusion with SDS, the kidneys were washed for 10 min with distilled water and then the organs were perfused for another 30 min with the SDS solution. After decellularization, the kidneys were perfused with distilled water for 1 h. Finally, decellularized organs were perfused for 1 h under sterile conditions with recirculated, sterilely distilled water containing 200 U/mL of penicillin and 200 μg/mL of streptomycin to prevent bacterial contamination.
Isolation and culture of primary human osteoblasts
Human osteoblasts were isolated from femur heads of patients undergoing prosthetic replacement, in accordance with the ethical code of “Klinikum rechts der Isar” (Technical University Munich, Germany) and having obtained the patients' written consent. Briefly, cancellous bone was removed mechanically from the femur head, washed three to five times with PBS followed by a 1-h incubation at 37°C with an equal volume of digestion buffer (PBS, 0.07% collagenase II (Biochrom)). After digestion, cancellous bone was washed with PBS and transferred to cell culture flasks in the presence of the culture medium (DMEM with 1 g/L glucose, supplemented with 20% FCS, 1% L-glutamine, 1% MEM vitamins, 2% HEPES buffer, 100 U/mL penicillin, 100 μg/mL streptomycin, and 285 μM L-ascorbate-2-phosphate). The medium was changed every 5 days. Within 2 weeks, the cells were growing out of the bone pieces. After reaching confluence, they were transferred to other cell culture flasks for further culture and expansion. 13
Seeding and cultivation of cells within the bioscaffold
Primary human osteoblasts in passage 3 to 7 were seeded into the decellularized kidneys through the vascular system. The phenotype of the cells was verified by alkaline phosphatase (ALP), collagen type I, and osteocalcin staining. Briefly, 107 cells were suspended in 10 mL of cell culture medium and injected into the renal artery of decellular kidneys through the attached catheter in two steps, 10 min apart. The seeded bioscaffolds were placed in sterile containers in a cell culture incubator under normal cell culture conditions (37°C, 5% CO2). Perfusion with a pressure of 50 mmHg was started 4 h after seeding to allow attachment of cells. The perfusion medium comprised the osteoblast growth medium (DMEM w/o calcium, supplemented with 20% FCS, 1% L-glutamine, 2% HEPES buffer, 100 U/mL penicillin, 100 μg/mL streptomycin, 285 μM L-ascorbate-2-phosphate, and 100 nM dexamethasone). Twenty-four hours after cell seeding, the perfusion pressure was increased to 100 mmHg and a total of 300 mL of cell culture medium was recirculated and changed every 3 days.
For coculture of HUVEC and primary osteoblasts, we proceeded similarly. In this study, a mixed suspension of 7×106 HUVEC and 3×106 primary osteoblasts was prepared in 10 mL of medium and applied through the renal artery, as described above. In case of coculture experiments, a 1:1 mixture of osteoblast growth medium (DMEM w/o calcium, supplemented with 20% FCS, 1% L-glutamine, 2% HEPES buffer, 100 U/mL penicillin, 100 μg/mL streptomycin, 285 μM L-ascorbate-2-phosphate, and 100 nM dexamethasone) and endothelial cell medium was used. Except for the mentioned differences, culture parameters were equal to the description above.
Visualization of the arterial system
To visualize the arterial system after decellularization, a red colored solution of Allura Red (Sigma) was injected using the connected catheter. The solution was produced by adding 10 μg of Allura Red to 20 mL of freshly made liquid solution of porcine gelatin in warm distilled water.
Histology
For histological investigations, bioscaffolds were cut transversally at the hilum into two parts, of which one was stored at −80°C and the other was fixed in 4% paraformaldehyde, embedded in paraffin and cut in slices with 3 μm in thickness with a rotary microtome. All samples were stained with hematoxylin and eosin. In addition, immunohistochemical stainings were performed. For immunohistochemistry, sections were deparaffinized and rehydrated, followed by endogenous peroxidase inactivation.
Immunohistochemistry was performed using the indirect detection method and Vectastain ABC avidin-biotin system (Vector Laboratories), combined with AEC substrate chromogen (Dako). Primary antibodies were used in the following concentrations: anti-Laminin 1:500 (Dako), anti-Fibronectin 1:250 (Dako), anti-Ki-67 1:500 (Millipore), anti-Vimentin 1:500 (Dako), anti-Osteocalcin 1:100 (Biotrend), anti-CD31 1:50 (Dako). As a negative control, isotype anti-IgG antibodies in corresponding dilutions were used. Counterstaining with hematoxylin was additionally performed.
ALP was detected on frozen sections 7 μm in thickness using the NBT/BCIP method (Roche Applied Science) according to the manufacturer's instructions. Costaining of CD31 and AP was performed on frozen sections.
Apoptosis was detected on paraformaldehyde-fixed paraffin-embedded (FFPE) tissue slices by using the nick-end-translation method, as previously described (Gorczyca et al.). 14 Slices treated with DNase I were used as positive controls.
SYBR Green staining was performed on FFPE tissue slices by using the SYBR Green I reagent at a concentration of 1:10000 in PBS.
RNA isolation, cDNA transcription, and real-time PCR
RNA was isolated from formalin-fixed paraffin-embedded tissue slices using the High Pure FFPE RNA Micro Kit (Roche Applied Science). RNA was transcribed into cDNA with the Quantitect Reverse Transcription Kit (Qiagen), following the protocol provided by the manufacturer. The cDNA was preamplified with TaqMan® PreAmp Master Mix Kit (Applied Biosystems) and subsequently qPCR was performed using TagMan® reagents (Applied Biosystems), both procedures being performed according to the manufacturer's instructions. Transcript levels of osteogenic-related genes were determined using the ready-to-use TaqMan Gene Expression Master Mix (Applied Biosystems). Measurements were performed in triplicate; a nontemplate blank served as a negative control. Amplification curves and gene expression were normalized to the housekeeping gene GAPDH. Primers for the genes COL1A1, BGLAP, SPARC, SPP1, TP53, PHEX, MEPE, PDPN, ALPL, RUNX2, SP7, and GAPDH were purchased from Applied Biosystems as ready-to-use primer mix as summarized in Table 1. The PCR program was set as follows: denaturation for 2 min at 50°C followed by 10 min at 95°C and amplification for 40 cycles. Each cycle included 15 s at 95°C and 60 s at 60°C. The device used was StepOnePlus (Life Technologies GmbH).
Data analysis of the quantitative PCR (qPCR) comprises comparing the amplification curve in the exponential phase of different targets with the amplification curve of a housekeeping gene, which serves as the internal control. The calculation method used in the present work was 2ΔΔCt using GAPDH as the reference gene. mRNA was taken as calibrator from the same cells that were used for recellularization experiments.
Statistics
Data are presented as mean±SEM. Statistical analysis was performed with GraphPad Prism (GraphPad Software, Inc.). The Kruskal–Wallis test was used as the nonparametric multiple group test. Statistical significance was reached at p<0.05. If significant, groups were compared with each other using the Mann–Whitney U test (p<0.017 after Bonferroni adjustment).
Results
Setup of the decellularization protocol
The first aim of the present study was to set up a standardized, time-efficient and valid procedure for perfusion decellularization of solid tissues. Freshly thawed rat kidneys were perfused with different decellularization protocols based on SDS detergent. Systematically, the duration of perfusion (0.5, 1, 2, and 4 h) and SDS concentrations (0.25%, 0.5%, 0.66%, and 1.0%) were varied to identify the fastest and most gentle protocol that safely leads to full decellularization under preservation of the general structure and integrity of the matrix. Microscopic investigation with HE staining and additional SybrGreen fluorescent staining revealed that perfusion times of 1h and more with SDS concentrations of 0.66% or 1% reproducibly resulted in complete decellularization (Fig. 1). Furthermore, no cell nucleus or remaining DNA could be detected in these groups with SybrGreen staining (Fig. 1).
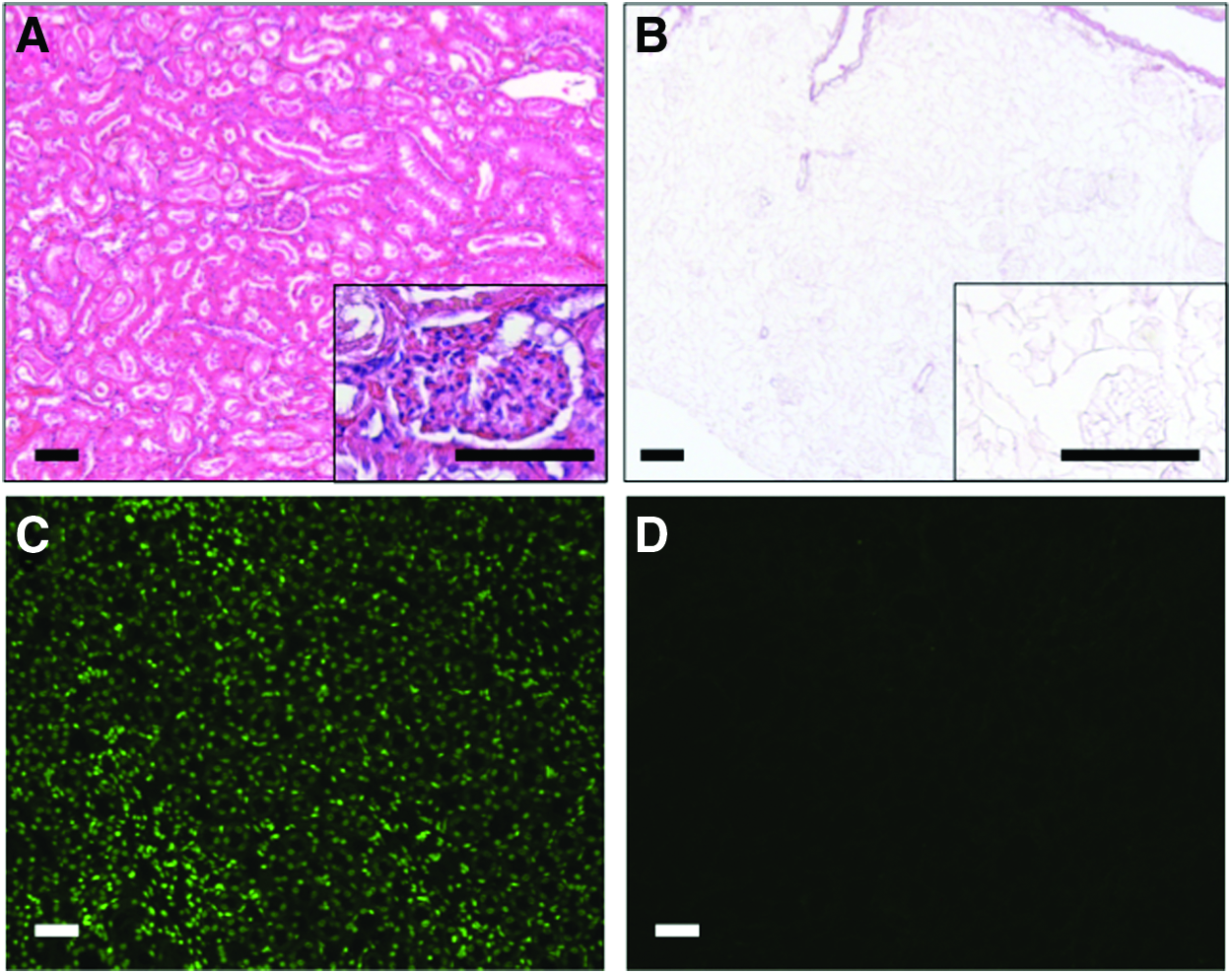
Decellularization of rat kidneys. Representative pictures of decellularized (0.66%, 1 h) versus native kidneys.
As it became clear that decellularization of 1 h with 0.66% of SDS is sufficient and leads to completely decellularized bioscaffolds from rat kidneys, this protocol was used for all further experiments.
Characterization of the decellularized bioscaffolds
During decellularization, the vascular network within the kidney appeared to remain macroscopically and functionally intact. This was indicated by persistent outflow from the renal vein and the ureter during and after the decellularization process. Thereby, no leaks appeared in the renal parenchyma.
The integrity of the vascular network was confirmed by visualization of the arterial branches by intra-arterial injection of Allura Red dye (Fig. 2). After injection of additional dye, the veins were also filled until the staining drained the renal vein.
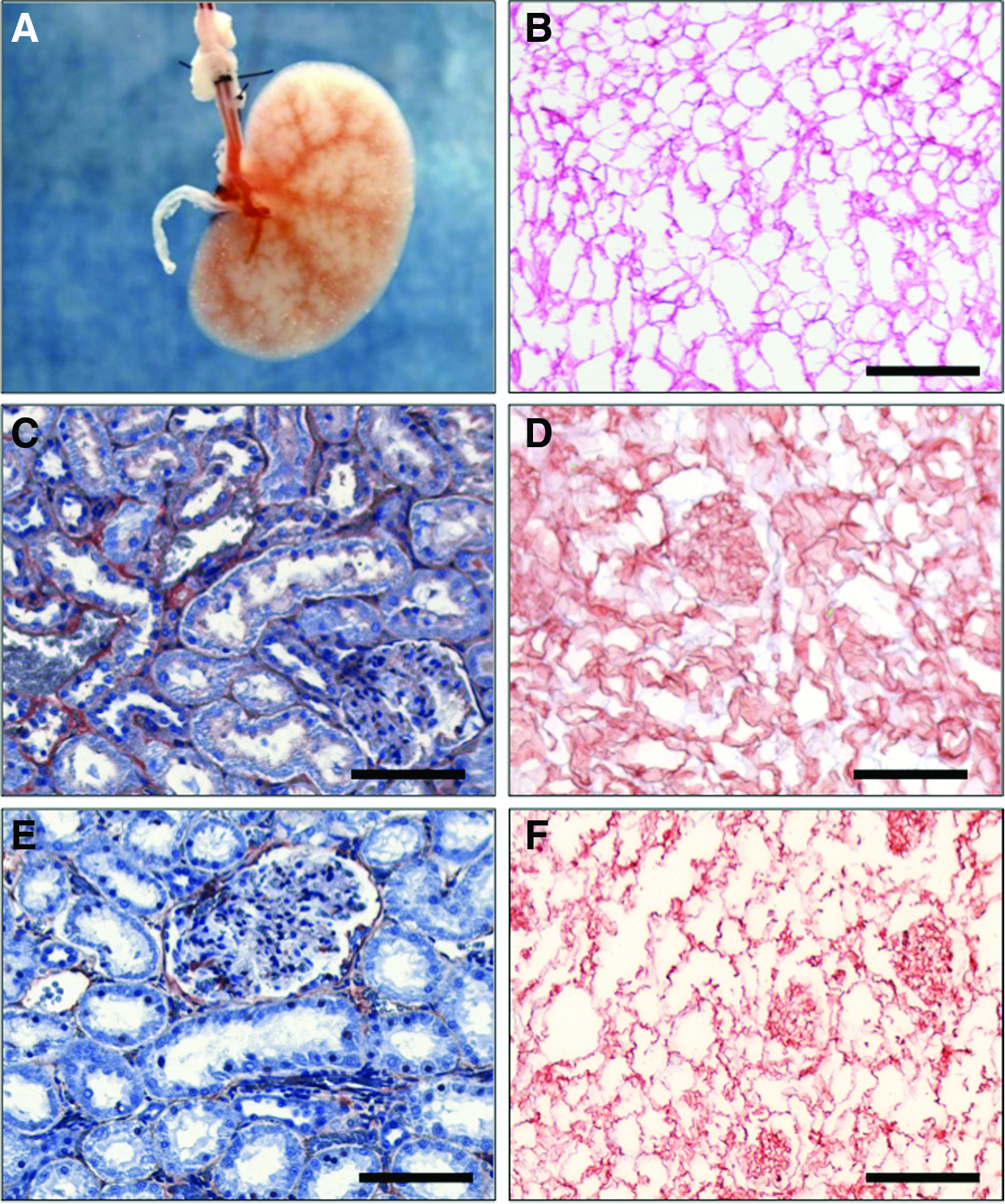
Characterization of decellularized kidneys. Representative pictures of decellularized (0.66%, 1 h) kidneys.
HE staining after decellularization shows a preserved architecture of the ECM with conserved glomerula and vessel structures in comparison to untreated kidneys (Fig. 1). Staining with Sirius Red reagent indicates that the remaining matrix is rich in intact collagen fibers (Fig. 2). Immunohistochemistry for laminin and fibronectin—two important components of the basal membrane—revealed that the integrity of the basal membrane remains preserved during the decellularization procedure (Fig. 2).
Distribution and survival of human osteoblasts seeded to the bioscaffold under dynamic conditions
The 107 primary human osteoblasts were seeded through the renal artery into each of the five independent decellularized kidneys. After 24 h of dynamic culture, the kidneys were harvested and HE staining was performed to investigate the distribution of the seeded cells. Osteoblasts were mainly detectable within the renal cortex. They appeared homogeneously distributed within the cortex around the larger vessels and in the glomerula (Fig. 3). HE staining was repeated after 5 and 14 days of dynamic culture on additional samples, each with five kidneys. Osteoblasts initially mainly located within the glomerula and around larger vessels increased in number and started to distribute over the whole cortex. Thereby, lumens of great vessels mainly remained free of cells (Fig. 3). Osteoblasts demonstrated a high proliferative activity over the whole period of investigation, as confirmed by strongly positive Ki67 immunostaining and expression of p53 in rtPCR at all points in time. In contrast, apoptosis within the seeded cell population remained constantly low over 2 weeks of culture, indicated by a low number of apoptosis-positive cells after 24 h, 5 and 14 days of dynamic culture (Fig. 3).
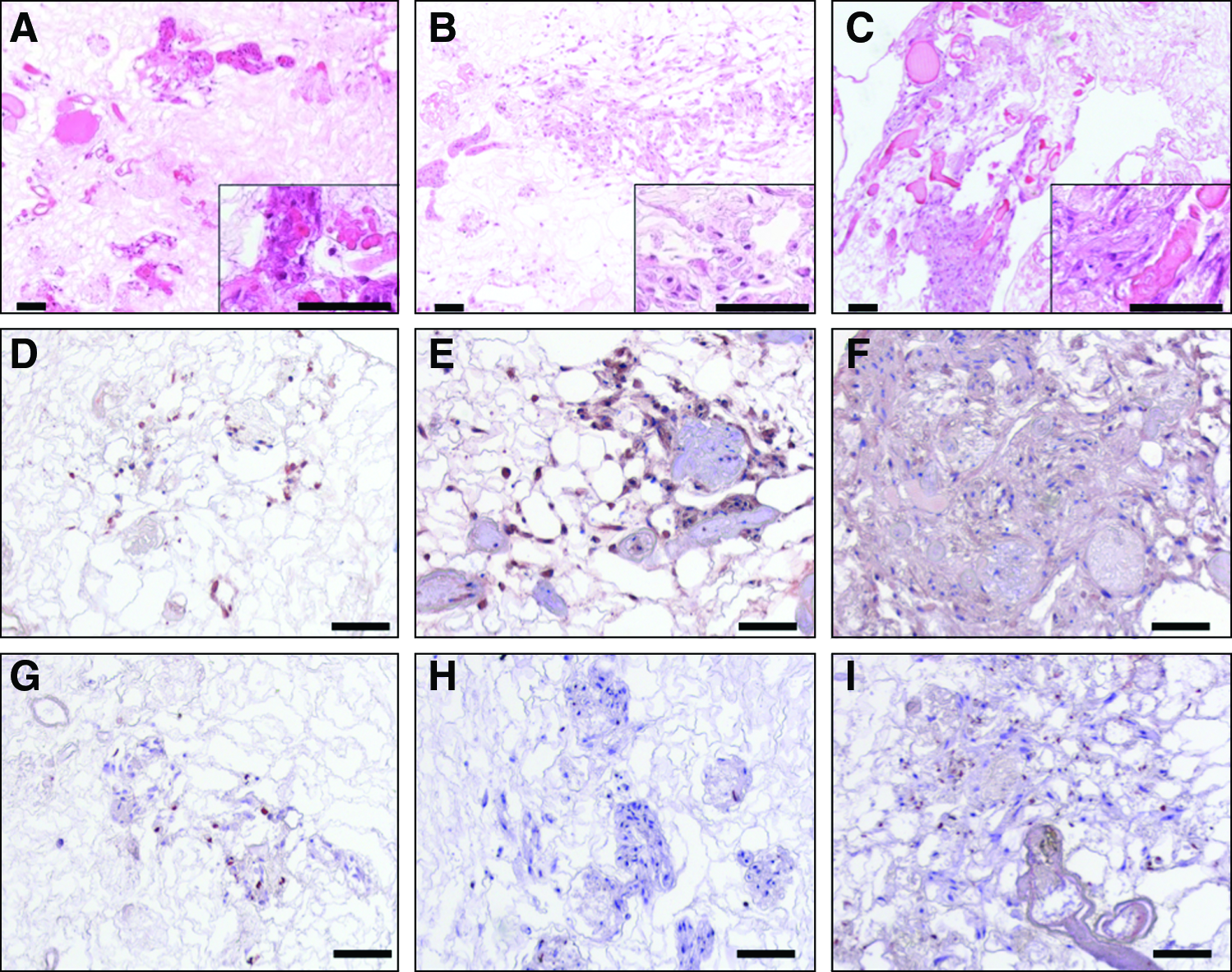
Decellularized kidneys seeded with human osteoblasts. Representative pictures of decellularized (0.66%, 1 h) kidneys seeded with 107 primary human osteoblasts.
Activity of the seeded human osteoblasts and remodeling of the ECM
To confirm that the human osteoblasts seeded on the decellularized rat kidneys retained their phenotype, we tested them for the expression of important bone markers by immunohistochemistry. Five independent samples were stained on day 1, 5, and 14 after seeding. Immunostaining of vimentin—a mesenchymal cell marker—showed robust detection of the protein over the whole culture period (Fig. 4). Histology for osteocalcin, one of the most bone-specific proteins of the ECM, revealed that large amounts of the protein were released into the biomatrix, where they accumulated over the observed culture period (Fig. 4). ALP staining was clearly positive for the seeded cells at all points in time. Interestingly, the intensity of the ALP activity peaked on day 1 and decreased toward day 14 (Fig. 4).

Characterization of human osteoblasts seeded into decellularized kidneys. Representative pictures of decellularized (0.66%, 1 h) kidneys seeded with 107 primary human osteoblasts.
rtPCR for important bone markers and transcription factors was performed on day 1, 5, and 14 of dynamic culture of three to four independent samples each. Seeded cells showed expression of the important osteoblast transcription factors Runx2 and osterix. Runx2 was expressed over the whole culture period, whereas osterix expression first appeared from day 5 onward (Fig. 5). Expression of the early osteoblast markers collagen type I and ALP decreased from day 1 onward. In contrast, the signals for the important bone matrix proteins osteopontin and osteonectin were detectable over the whole culture period. Analysis of the osteocyte markers PHEX and podoplanin revealed a strong expression of these factors until day 14 (Fig. 5).

Gene expression of human osteoblasts seeded into decellularized kidneys. rtPCR of primary human osteoblasts seeded into decellularized (0.66%, 1 h) kidneys. The nonparametric Kruskal–Wallis test for multiple groups revealed significant differences in expression between day 1, 5, and 14 in all tested gens (p<0.05). The Mann–Whitney U test was used to compare expression of day 5 and 14 to day 1. The Mann–Whitney U test showed significance in gene expression in some markers (*p<0.017). n=3 (1 and 5 days) and n=4 (14 days); each kidney is measured in triplets.
Coculture of primary human osteoblasts and endothelial cells within bioscaffolds
Bioscaffolds were seeded with a suspension of 7×106 HUVEC and 3×107 primary human osteoblasts through the renal artery. Seeding was performed in three independent experiments into a separate bioscaffold each. After 5 days of dynamic culture, seeded bioscaffolds were investigated by histology. HE staining revealed a comparable distribution pattern of the cells within the biomatrix, as seen in further experiments after seeding osteoblasts only. In this study, CD31 immunohistology revealed that HUVEC were mainly located within the vessels, where they lined along the walls. In contrast, osteoblasts were preferentially detectable in the matrix around the vessels. The latter appeared strongly positive in AP staining, even though if they were growing in direct contact to HUVEC (Fig. 6).
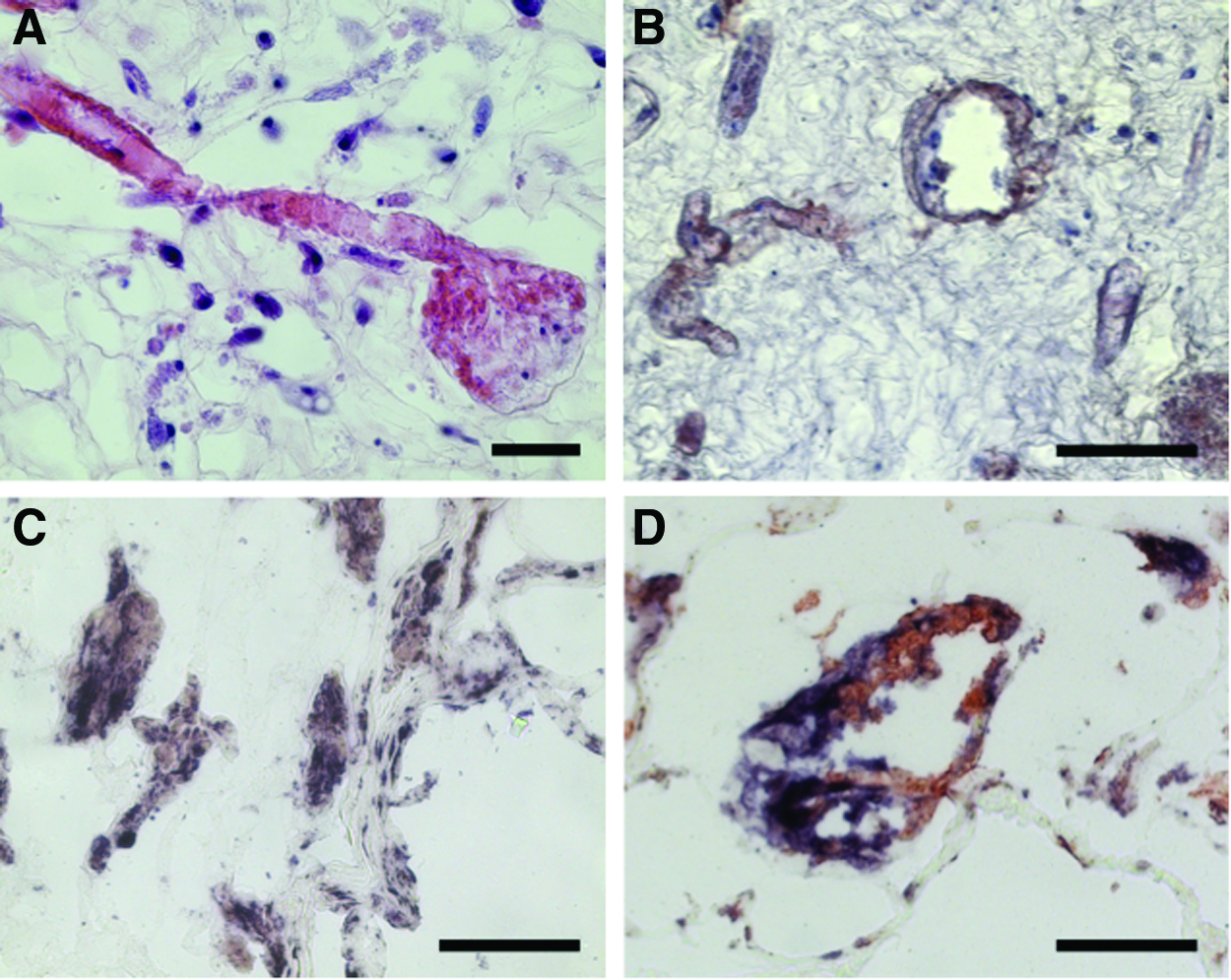
Coculture of primary human osteoblasts and endothelial cells. Representative pictures of decellularized (0.66%, 1 h) kidneys seeded with a mixture of 7×106 human umbilical vein endothelial cells (HUVEC) and 3×106 primary human osteoblasts.
Discussion
Within the present work, we could extend the relevance of bioscaffolds as a basis for tissue engineering of vascularized tissues. Our study confirms that bioscaffolds are capable of growing cells under dynamic conditions, independently of their tissue and even of their species. Furthermore, we could demonstrate that osteoblasts seeded on a bioscaffold change the composition of the matrix toward their natural niche by, for example, producing collagen, osteocalcin, osteonectin, and osteopontin. Therefore, the decellularization technique may have the potential to become a platform technology for tissue engineering, as it offers a universally applicable and easily producible scaffold that addresses the yet unsolved problem of vascularization.
As shown in the present work—and particularly by the previous work on implanting bioscaffold-based constructs in vivo—the concept of decellularization provides a highly demanded solution for the challenge of vascularization in tissue engineering.6–8,15 The technique provides a scaffold with an inherent physiologically shaped vascular network, allowing perfusion of the matrix. Recent work proved that SDS-azellularized kidney biomatrices can be re-endothelialized in vitro and be subsequently reimplanted in vivo. 15 A sufficient vascular supply is the critical prerequisite to realize tissue engineering of tissue constructs of more than 400 μm in diameter. 1 Alternative approaches with artificial matrices that provide an inherent vascular network have failed up to now. 1
In the first part of the present study, we demonstrate a standardized procedure for time-efficient decellularization of solid tissues. The combination of physical cell lysis by freezing and subsequent thawing with perfusion with the detergent SDS leads to full and reproducible decellularization of rat kidneys with only 1 h of SDS perfusion. Thus, a ready to use biomatrix can be available from a frozen rat kidney within 5 h. This is significantly faster than previously published protocols, lasting between 1 and 10 days.4–6,10,15,16
For decellularization, we used the relatively strong detergent SDS that was already successfully used for decellularization of avascular tissues in our department. 17 We applied the decellularization agent by perfusion through the vascular system. This is critical, as the immersion of a solid tissue only leads to decellularization of tissues up to 5 mm in thickness. 18 We decided to use SDS, as previous studies confirmed the superior decellularization activity of SDS compared with milder detergents, thus making it the best candidate for fast and complete decellularization.5,10 However, in high concentrations and long incubation periods, the strong detergent SDS can also lead to the destruction of the ECM. 19 Thus, we investigated the remaining matrix after decellularization of the kidneys with the presented protocol. In this study, we could confirm that the architecture of the ECM with the vascular network remains intact. Furthermore, even the integrity of the basal membrane is preserved, which is a critical prerequisite for further endothelialization of the vessels. 10
To obtain a universally applicable biomatrix, a partial denaturation is even favored to eliminate tissue-specific signals from the matrix that may be immunogenic. This is necessary to avoid incalculable influences on the cells seeded later. The native ECM contains a variety of signals mediated by integrins and cytokines that regulate differentiation and proliferation of the residing cells. 9 Ross et al. showed that this ability is still present in the ECM of rat kidneys decellularized by Triton X-100, leading to terminal differentiation of pluripotent stem cells seeded into the biomatrix. 10 Here, the use of the stronger denaturating SDS provides advantages over milder detergents, as it further reduces tissue-specific signals. 20
In our experimental setting, we used rat kidneys as scaffold source and seeded human osteoblasts. The use of human cells makes the model relevant for a variety of in vitro models and also for potential therapeutic scenarios.
Although osteoblasts are relatively large cells, seeding through the vascular network resulted in homogeneous distribution of the cells within the scaffold without damaging the matrix. In contrast, we were not able to achieve a homogeneous seeding of human osteoblasts using the ureter in our pretests. In this study, cells were mainly remaining in the renal calyx and the distal renal tubuli (data not shown). The reason may be that human primary osteoblasts are too large to sufficiently ascend the renal tubular system. Nonetheless, seeding through the ureter is possibly promising for future applications, using smaller cell types. This would allow direct seeding of cells into the extravascular compartment. 15
Seeding through perfusion is a great advantage over alternative seeding strategies. If cells are just seeded onto the surface of the decellularized solid tissue, they have to migrate from there toward the core of the scaffold to colonize the matrix. Here, a homogeneous distribution of the cells is not possible, especially if the seeded cells are large and thus slowly migrating. 21 An alternative approach is the direct injection of cells into the matrix resulting in inhomogeneous and irreproducible distribution of cells and damaging of the scaffold. 5
Human osteoblasts seeded into the rat kidney-derived biomatrix retain their phenotype for at least 2 weeks of culture in the presence of the standard osteogenic medium without addition of cytokines. The investigation of their expression profile revealed that they did not only maintain their osteoblast phenotype, they even maturated toward a more osteocyte-like phenotype. During the culture period, the expression of early bone markers ALP and collagen type I decreased, whereas the expression of late bone markers osterix, osteonectin, and osteopontin, as well as osteocyte markers Phex and podoplanin remained high or even increased. This is remarkable, as the remaining matrix of decellularized tissues can contain signals from the former tissue, leading to potential dedifferentiation of the seeded cells. 10 Histology of the scaffolds, 14 days after seeding the osteoblasts, revealed that they remodeled the biomatrix more toward the bone niche by releasing high amounts of osteocalcin-containing ECM.
Coculture experiments with endothelial cells indicated that the latter do not negatively influence differentiation of the cultured osteoblasts within the matrix. Furthermore, endothelial cells were predominantly located within the vessels, as osteoblasts were detectable in the surrounding matrix. In line with further investigations, seeded endothelial cells began to re-endothelialize the bioscaffolds under dynamic culture conditions.6,8,15 This is a critical finding, as sufficient endothelialization is a basic requirement to implant a tissue-engineered construct into an organism. 1
It is known that the 3D structure of the ECM also influences differentiation and proliferation of cells.22,23 It is likely that the seeded cells also adapt the 3D structure of the bioscaffolds, but this needs further investigation and above all longer observation periods to be proven. If stem cells seeded on a bioscaffold can also be reproducibly differentiated into cell types from a tissue other than the scaffold's origin remains unclear. Furthermore, it has to be shown if they are able to remodel the matrix toward another tissue. In the future, it will be important to answer these questions to further evaluate the significance of bioscaffolds for tissue engineering applications.
Although the results from the present study are encouraging, they have some limitations. For future clinical application, a construct larger than a rat kidney may be necessary to treat large bone defects. It remains unclear, if the presented findings are also valid for larger bioscaffolds from other species or tissues. Furthermore, the used technique for culture of the seeded bioscaffolds was too susceptible for contamination. During the study, several constructs were lost due to contamination. Here, improvements in the bioreactor design will be necessary for future experiments.
Footnotes
Disclosure Statement
No competing financial interests exist.