
Abstract
Successful cell replacement therapy in the central nervous system (CNS) depends on both the transplanted cell type and the cell delivery method. It was established that differentiated neurons are the most desirable cell source; however, they are highly sensitive to dissociation shear; removing them from the growth surface inflicts serious damage, rendering them less viable for transplantation. Pilot experiments using glass colloids as injectable cell carriers for cell transplantation in the adult rat hippocampus have greatly improved neuron survival and long-term neuron integration. However, these early studies have highlighted glass particle shortcomings. They are uncompressible, and, thus, only a small number of beads can be injected, limiting the transplanted cell number. Moreover, they remain permanently in the brain. To improve colloidal carriers properties for cell transplantation and establish a basis for the next generation of cell delivery supports, we have designed a broadly applicable engineering strategy to enable neuronal cell growth on and release from hydrogel particles before transplantation. Here, we describe poly(N-isopropylacrylamide) (pNIPAM) particle preparation, and we demonstrate that these hydrogel particles both facilitate manipulation of neurons and enable the increase in the number of viable transplanted cells in the young adult rat hippocampus. The absence of long-term cell association to beads suggested that pNIPAM thermoswitching properties enable the separation of cells from the beads during injection, which minimizes the number of injected carriers. Contrary to observations with glass carriers, no particle clumping was observed at the injection site, which indicates minimal risk of long-term inflammatory responses. Taken together, the properties of hydrogel particles make them a promising micro-carrier to improve neuronal cell transplantation.
Introduction
S
A new generation of microgel particles (MGPs) derived from an injectable polymerizable hydrogel 26 has emerged from the recent development in microfluidics platform.27–32 Neuronal progenitor cells can be encapsulated in hydrogel, enabling protection from the host immune response, yet providing a microenvironment that fosters cell differentiation. However, as cells differentiate, neuronal processes get entangled with the polymer mesh, 33 impeding cell migration and decreasing the probability that transplanted cells make a functional connection with the host circuit. To permit cell dispersion and facilitate functional insertion into the host circuit, cells should be grown on the surface of the MGPs, and the particle surface should be engineered with the dual abilities of fulfilling neuronal cell growth specific requirements and enabling controlled release from the particle surface.
Stimuli-responsive polymers are known for their changing physical properties on trigger. Among the hydrogels used in biomedical applications, poly(N-isopropylacrylamide) (pNIPAM) reversibly swells or shrinks in response to temperature changes between room temperature, 20°C and body temperature, 37°C. This thermoswitching property has attracted substantial interest for a wide range of functional applications.34–38 Furthermore, pNIPAM exhibits low nonspecific protein adsorption, making it a material of choice for applications such as protein-ligand recognition, 39 artificial organ transplantation, 40 and targeting of cell culture surfaces to control cell release from two-dimensional (2D) cell culture surfaces.41,42 While large pNIPAM particles can be produced with the desired thermoswitching sensitivity in a temperature range compatible with neuronal cell physiology, 27 the particle native surface does not allow neuronal cell adhesion and growth. If the pNIPAM particles could be engineered to support neuronal cell culture, they could replace glass carriers by incorporating the advantageous properties of hydrogels with colloidal carriers for use in transplantation. On this basis, we developed a pNIPAM particle fabrication protocol aimed at preserving thermoswitching properties for cell release while enabling neuronal cell differentiation, and we compared their cell delivery abilities with those of the previously used glass particles.
Materials and Methods
The particle fabrication process can be broken down into three steps detailed next.
MGP production by emulsion-templated polymerization
To form MGPs by emulsion-templated polymerization, an aqueous phase containing N-isopropylacrylamide (NIPAM) monomers and N,N′-(1,2-Dihydroxyethylene)-bisacrylamide cross-linkers with ammonium persulfate as the initiator was emulsified in dodecane (Fluka) in a co-flow microfluidic platform using span80 (Fluka) as the stabilizing surfactant following published parameters.27,43–46 Polymerization accelerator N,N,N′,N′-Tetramethyl-ethylenediamine was added to the continuous phase downstream from where droplets formed. The accelerator diffused in the droplet and catalyzed radical polymerization. Droplets were kept rolling in the microchannel during polymerization. The final particles used in transplantation were collected by sedimentation in water. To minimize the loss of MGPs by nonspecific adhesion, all plasticware, tubes, and pipets tips used to handle the particles were silanized by vapor deposition of hexamethyldisilazane. Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich.
To remove all traces of polymerization reagents that are toxic to cells, such as oil and detergents, particles were extensively washed in isopropanol (IPA), which is a good solvent for the chemicals used. IPA was replaced thrice before transferring the MGPs to distilled water for long-term storage. Before their use for cell culture, MGPs were sterilized in 70% ethanol overnight at room temperature. To maintain a sterile environment, all subsequent steps were carried out in a biosafety cabinet with solutions filtered through a 0.22 μm filter (0.20 μm sterile syringe filter, 28 mm syringe filters, Corning, Inc.).
Microgel characterization
To estimate the size distribution of the MGPs produced in co-flow device, we measured the diameter of more than 500 particles at room temperature (RT). To obtain the best detection of the edge of the particles, images of the MGP equatorial plane were acquired with a 10X phase-contrast objective. Images were processed in ImageJ (Fiji, Vers. 1.43 h). We measured the intensity profile along a line passing through the center of the MGP. The particle diameter was determined by the peak-to-peak distance.
MGPs thermoswitching properties before and after layer-by-layer deposition were evaluated by measuring the change in particle diameter as the temperature was continuously changed from RT to 40°C using a temperature controller (Warner Instrument Corporation) mounted on the microscope. To ensure that the maximal size had been reached, the sample was maintained at 40°C for 5 min, before switching off the heating platform and letting the sample return to RT. Images were acquired in 30-s time intervals using the acquisition software of the microscope (NIS Elements; Nikon). The particle size was measured offline in ImageJ.
The elastic properties of the MGPs were measured by atomic force microscopy (AFM; Nanowizard® BioScience AFM, JPK Instruments AG, mounted on an Axiovert 200M inverted microscope; Zeiss) at 25°C and 37°C. Nonconductive silicon nitride cantilevers with force constants between 50 mN/m and 120 mN/m were used (model MLCT and NP-O10; Veeco Instruments, Inc.). A spherical indentor consisting of a 10 μm glass bead (Gerlinde) was glued to the cantilever using Araldite® Rapid (Huntsman Advanced Materials). To obtain a reproducible and quantitative measure of the elastic properties of a particle, the spherical indentor was brought in and out of contact with the particle one thousand times. The resulting forces curved measured were fitted using a Hertzian model to determine the Young's modulus using a Poisson ratio of 0.45. 47
Microgel surface coating for neuronal cell culture
To enable electrostatic deposition of poly-L-lysine (PLL, Mw=300.000 g/mol; Sigma-Aldrich), which is necessary for neuronal cell adhesion, we used a layer-by-layer deposition of positively and negatively charged polyelectrolytes to increase the surface charges of the MGPs. This process was adapted from the shell particle fabrication protocol described by Radtchenko et al. 48
The first layer was deposited by incubating the MGPs in poly(allylaminehydrochloride) solution (PAH, Mw=8000–11,000 g/mol, Sigma-Aldrich, dissolved at 5 mg/mL in the potassium acetate), for 25 min on a rolling shaker. Next, the excess PAH was removed by washing the particles thrice with distilled water. The next counter ion layer was deposited by incubating the MGPs in poly(sodium-4-styrene-sulfonate) solution (PSS, Mw=70,000 g/mol, Sigma-Aldrich, 5 mg/mL in the potassium acetate) for 25 min on a rolling shaker. Next, the excess PSS was removed by washing the particles thrice with distilled water. The PAH/PSS deposition cycle was repeated at least thrice to obtain the desired negatively charged polymer shell that enables electrostatic adsorption of the amount of PLL necessary to obtain a robust neuronal cell adhesion on all the MGPs. At this stage, the PAH/PSS-coated particles could be stored in water at 4°C for at least 2 weeks without appreciable changes in their properties.
The final PLL coating step was performed when particles were required for cell culture. The MGPs were left incubating overnight on a rolling shaker in a PLL solution (5 mg/mL in pH 7.4 PBS). The excess PLL solution was removed, and the particles were then washed thrice with PBS. Twelve hours before cell seeding, PLL-coated MGPs were incubated in culture medium at 37°C to enable the medium to fully penetrate the microgel.
To estimate the increase in adsorbed PLL when we increased the number of deposition cycle, we fluorescently labeled the accessible primary amine of the deposited PLL. After each new deposition cycle, a small amount of particles was set aside and was coated with PLL. The PLL-coated MGPs were then suspended in 0.2 M borate buffer (pH 7.4) and left to equilibrate for at least 15 min. The labeling reaction was started by adding Alexa 555-NHS ester (Life Technologies; 10 mg/mL in dimethyl sulfoxide), a small diffusible fluorescent dye carrying a reactive succinimidyl ester group, to the MGPs suspension (ratio 1:50 v/v). The MGPs were kept on a rolling shaker for 1 h in the dark. The labeling reaction was stopped by adding hydroxylamine hydrochloride (Sigma-Aldrich; 1.5 M at pH 8.5) to the reaction solution (ratio 1:10 v/v). After 30 min, the particles were washed thrice with distilled water. The number of labeled primary amine correlated with PLL surface concentration. Hence, the measure of the residual fluorescence provided a relative measure of PLL deposited as a function of the number of deposition cycle. The cross-section of the labeled MGPs were imaged by confocal microscopy. To enable the comparison of the fluorescence intensities between samples, we kept the acquisition parameters constant (identical laser power, scanning rate, and detector gain) and we imaged them on the same day. For each particle, we measured the averaged radial intensity profile. The amount of deposited PLL for a given number of deposition cycles was determined by averaging the mean radial intensity profile of at least 10 particles.
Glass bead preparation
Borosilicate glass beads of 45 μm size (MO-SCI Specialty Products) were sterilized in a 70% ethanol solution overnight. Dry beads were incubated in PLL (Mw=300,000 g/mol; Sigma-Aldrich) 0.2 mg/mL solution overnight. PLL solution was aspirated, and beads were washed with cell culture grade water thrice for 40 min. Dry beads were distributed in a cell culture dish before adding medium and cells.
Neuronal cell culture
Hippocampal neuronal cells from embryonic rats were harvested at 18 days postfertilization (embryonic day 18, E18) following standard procedures. The cells were seeded in a 12-well plate (Greiner Bio-One) at a density of 100,000 cells per well. Each well contained a thin layer of coated particles pre-equilibrated in cell culture medium. The pNIPAM particles were slightly denser than water and, thus, settled at the bottom of the dish. However, they were not as dense as glass particles, and, thus, they required extra care not to aspirate MGPs during liquid handling and washing steps. Furthermore, all the solutions had to be prewarmed to minimize undesirable thermoswitching. The medium was composed of Neurobasal-A (Gibco) with Serum-Free Supplement B-27 (1 mL per 50 mL), 200 mM L-glutamine, 2 units/mL of penicillin, and 2 μg/mL of streptomycin. No additional serum or growth factor was added to the cell culture unless otherwise specified. All cell culture reagents were purchased from Life Technologies. Culture growth was assessed each day by means of bright field microscopy (Axiovert 25 microscope, Zeiss and ProgRes C10plus camera).
Measure of the total DNA for the glass and the MGP cell cultures
The same procedure was used for both cell carrier types. At DIV5, the cell culture medium was removed and the cultures were washed with 500 μL Hank buffer. The harvested cells were lysed, and DNA was extracted using standard DNA extraction procedure (Zymo Research kit). The final concentration was determined on a NanoDrop spectrophotometer device (Thermo scientific) by measuring the absorbance at 260 nm wavelength.
Cell transplantation
E18 neuronal cell cultures were grown on MGPs for 5 days. Cells were infected at 2 days in vitro (DIV 2) with lentiviral particles to express GFP under the synapsin promoter to specifically label neurons. The medium was conditioned by replacing a third of the culture with medium harvested from 2-week-old E18 hippocampal neurons that had been grown in 2D conditions. After 48 h, the culture was washed with HBSS to remove remaining traces of the lentiviral particles, and fresh conditioned medium was then applied. At DIV 5 and 6, microgels bearing neurons were uptaken into a syringe at RT. The combination of the temperature change and the shear applied while loading the needle caused the cells to separate from the MGP, and the MGPs without cell load stuck to accessible surfaces. As a consequence, solely dissociated cells were stereotaxically injected into the hippocampus of the brain of adult female Fischer 344 rats (120–150 g, 6 weeks old). The procedure was performed on both right and left hippocampus. Coordinates for the injection were taken from Paxinos and Watson adult rat brain atlas. Coordinates in millimeters: (anteroposterior [AP], −3.5; mediolateral [ML],±3.0; dorsoventral [DV], −3.9) for CA3 injections and (anterio-posterior [AP], −3.5; mediolateral [ML],±1.15; dorsoventral [DV], −4.1) for dentate gyrus (DG) injections. The animals were deeply anesthetized with a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg) before the injection, and 0.5 μL of the bead suspension was injected with a 26-gauge beveled needle mounted on a 2 μL Hamilton syringe. All procedures were carried in accordance with the “Guide for the Care and Use of Laboratory Animals.”
Animals were sacrificed at different time points post-transplantation. Rats were anesthetized and transcardially perfused with 0.9% (w/v) saline followed by 4% paraformaldehyde in phosphate-buffered saline. Brains were excised, postfixed in 4% paraformaldehyde overnight at 4°C, and stored in 30% sucrose for cryoprotection before sectioning. Sequential 45 μm coronal sections were prepared, and the resulting sections were immuno-stained to assess the implantation of injected neurons by quantifying GFP expression by microscopy.
Immunostaining
All cell culture samples were fixed and stained using standard immunocytochemistry protocols. The following primary antibodies were used: mouse anti-Nestin monoclonal (1:700); mouse anti-NeuN (neuronal nuclei) monoclonal (1:2000); mouse anti-GFAP monolyclonal (1:1000); and mouse anti-Smi-312 monoclonal (1:500). All primary antibodies were purchased from Chemicon Millipore. All cultures were double stained with rabbit anti-Tuj-1 (neuronal class III β tubulin) monoclonal (1:1000), from Hiss Covance. Secondary antibodies were used as follows: goat anti-rabbit IgG alexa 555 and goat anti-mouse IgG alexa 488. Both secondary antibodies were diluted at 1:500, and they were purchased from Invitrogen. DAPI was used to stain the nuclei, and it was diluted 1:1000 in distilled water.
Brain slices were fixed and stained using standard protocol for immunohistochemistry. Signals from GFP+cells were amplified with primary rabbit anti-GFP and the corresponding secondary antibodies (labeled with Alexa Fluor 488). To highlight the DG, rabbit anti-Prox-1 (1:200) from Chemicon Millipore was used. Finally, some sections were stained with Nissl-red (Invitrogen) to identify the neuronal cell nucleus.
Sample preparation for scanning electron microscopy
Since a colloidal suspension cannot be dehydrated in a critical point dryer without losing the sample, we developed a scheme to tether colloidal particles to a coverslip using the neuron's natural ability to grow processes on permissive surfaces while searching for cognate partners. At DIV 2 or 5, particles carrying neuronal cells were transferred to PLL-coated coverslips typically used for neuronal cell culture. Neurons growing on the particles extended processes on the coverslips, which physically tethered the particles and provided sufficient mechanical stability to withstand all the preparative steps. After 36 h, samples were gently washed with PBS before fixation overnight in a 2% glutaraldehyde solution. Coverslips were then dehydrated by successive treatment with incremental acetone concentrations to approximately 100% and then dried in a critical point drier. The dry coverslips were transferred to a sputter coater where a thin layer of gold was deposited over the samples. Samples were stored in a dust-free environment until image acquisition on a Hitachi TM 1000 (Acceleration voltage 15 kV, BSE-detector). The same protocol was used for both glass and MGPs.
Results
Microgel pNIPAM particles (MGPs) were produced using an inverted emulsion as a polymerization reactor. The emulsification process was carried out in a co-flow microfluidics device to control the particle size and ensure uniform polymerization of the microgel (Fig. 1a).27,43–46 To remove all traces of the reagents, particles were washed extensively and then stored in filtered water at 4°C in their native state. Under these storage conditions, the pNIPAM particles remained stable for more than 24 months. The cross-linked polymeric network retained the shape of the droplet, leading to thermoswitching particles with a relatively uniform diameter of 120±15 μm at 20°C (50±3 μm at 37°C) (Fig. 1b–d). The elastic modulus of these particles measured by AFM indentation ranged from E=21.4±0.5 kPa at 25°C to E=65.1±6.4 kPa at 37°C close to the elastic modulus measured for the CNS, which ranges between 0.1 and about 1.0 kPa depending on the animal age and the area of the brain probed.49,50
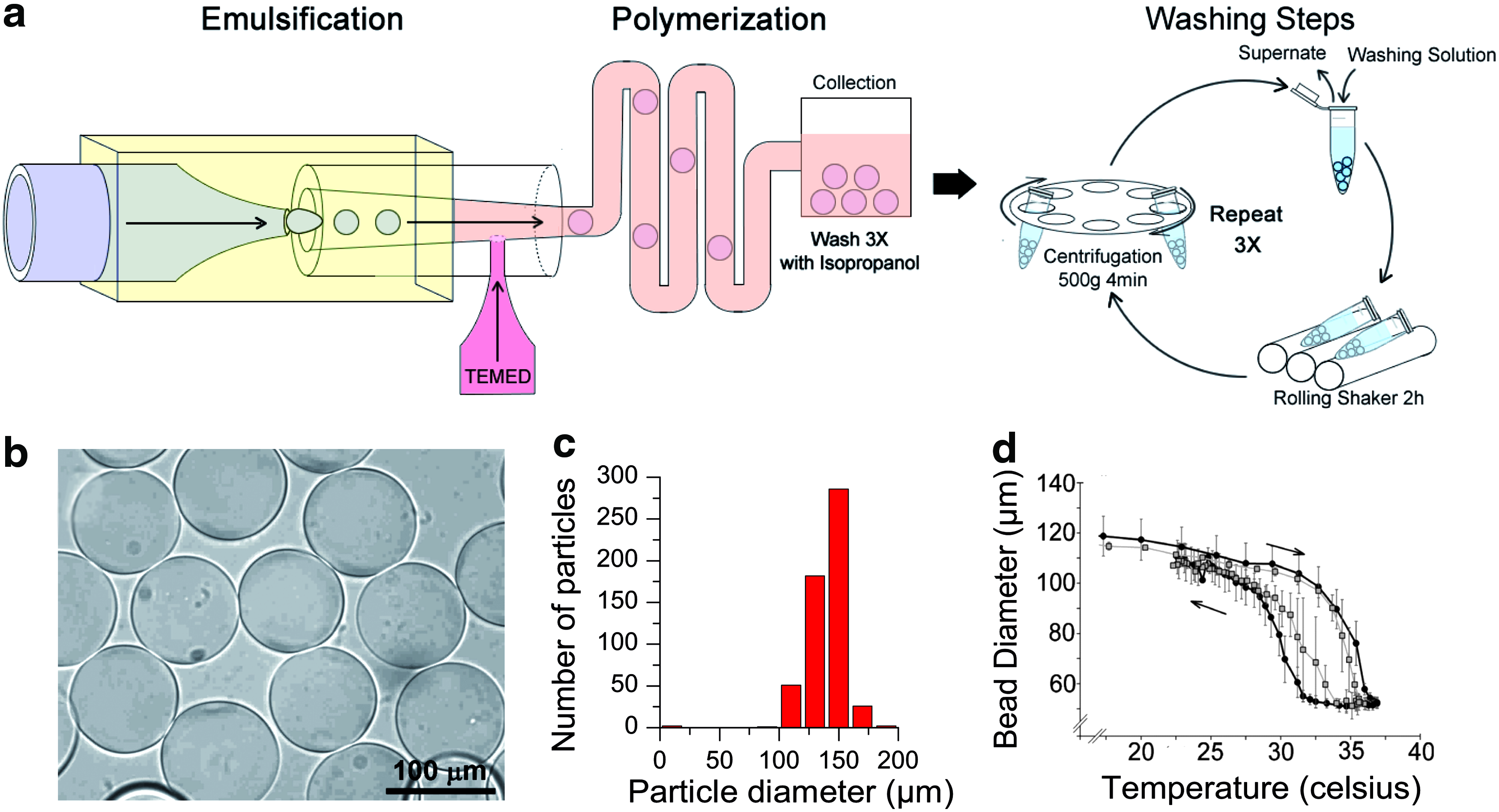
Poly(N-isopropylacrylamide) (pNIPAM) particle prepared in microfluidics device.
The surface properties of the native MGPs were not suitable for neuronal cell culture. Rat primary hippocampal cells preferred forming free floating cell clusters rather than adhering to the native uncharged MGPs (Fig. 2a), and pNIPAM particles required further processing to support neuron cell growth. Negatively charged glass surfaces are typically coated with PLL 51 to promote neuronal cell adhesion. Thus, we devised a noninvasive layer-by-layer deposition of polyelectrolytes 48 schematically depicted in Figure 2b to form an adhesive PLL shell on the MGPs. This method presents the advantage to be independent of the underlying polymer network and, thus, circumvents the need to engineer functional reactive sites for covalent cross-linking of adhesion peptides that could alter the pNIPAM physical properties. The deposition of PLL was visualized by including 1% fluorescently labeled PLL to the PLL coating solution. Confocal images of the particle cross-sections showed a thin fluorescent layer around the particles (Fig. 2c). The absence of FITC-PLL penetration within the hydrogel indicated that the polymeric (PAH/PSS) shell restricted diffusion of large molecules into the hydrogel.

pNIPAM particles coating
To estimate the increase in adsorbed PLL when we increased the number of deposition cycle, we labeled PLL accessible primary amines using a small diffusible fluorescent dye carrying a reactive succinimidyl ester group (Materials and Methods section). The MGPs were extensively washed to remove noncross-linked dye before we visualized the dye cross-linked to the adsorbed PLL by laser scanning confocal microscopy (Fig. 2d). We measured the radial fluorescence intensity profile for each particle. The mean radial intensity profiles for a given number of deposition cycle was obtained by averaging the radial intensity profiles of at least 10 particles (Fig. 2e). With each deposition cycle, the mean fluorescence intensity of the particle surface increased, indicating an increase in PLL surface concentration. A minimum of three deposition cycles of PAH/PSS polyelectrolytes were necessary to observe a robust neuronal cell adhesion and neuron differentiation in all MGPs, indicating that the critical PLL surface concentration had been reached. Measurement of coated pNIPAM particles' thermoswitching properties between 18°C and 37°C showed similar amplitudes as the native particles during swelling after a temperature decrease and deswelling after a temperature increase (Fig. 1d, open circle). The only appreciable effect of the polymeric shell was to reduce the thermoswitching hysteresis. These results confirmed that this approach forms a uniform PLL coating while preserving the physical properties of MGPs.
We next examined neuronal cell growth and differentiation on PLL-coated MGPs. Primary E18 hippocampal cells were seeded on MGPs pre-equilibrated in culture medium at 37°C for 12 h. Within a few hours, seeded cells adhered to the MGPs (Fig. 3a). After 3 days, cell bodies covered the MGPs. Rarely, when cells were directly on top of the particle, the morphology of growing neurons could be discerned with more cells out of focus bridging two particles (arrow, Fig. 3b). During the first week in culture, neuron processes were not dense enough to fully encage the MGPs. Hence, changes in particle diameter caused by changes in temperature during sample handling could lead to the separation of the cells from their carrier as seen during imaging (Fig. 3c). To visualize specifically neurons, cells were infected using lentivirus to express a cytosolic fluorescent protein, Tandem-dimer-Tomato (TdT), under-synapsin promoter. Real-time confocal imaging of the 3-day-old culture at 37°C showed that the FITC-PLL layer deposited on the MGPs was still present (Fig. 3d), and that some TdT-labeled neuronal processes were running over the particle surface (Fig. 3e). The corresponding transmission image showed that more TdT-negative cells were present on the bead surface corresponding to undifferentiated neuronal cells (Fig. 3f). To promote a complete neuron differentiation, one volume of medium harvested from a mature, 2-week-old neuron culture was added to one volume of cell culture medium to all the subsequent culture. Immuno-cytochemistry performed on these cultures after 4 days in vitro (DIV 4) showed cells positive for both tuj-1 early neuronal marker and smi-312, an axonal marker, confirming that embryonic neurons differentiated and polarized (Fig. 3g–i); while a small fraction of the cells was still nestin positive (Fig. 3j, k). Stains for glia cells marker GFAP were negative (Fig. 3l, m). Reconstruction of three dimensional confocal imaging suggested that processes ran on the surface of the microgel and did not penetrate the polymer matrix. At the time of transplantation (DIV 5), the composition of the cell population measured for MGP carriers was within the standard deviation of the population measured for glass carriers (Table 1), suggesting normal neuronal development of soft pNIPAM carriers in conditioned medium.
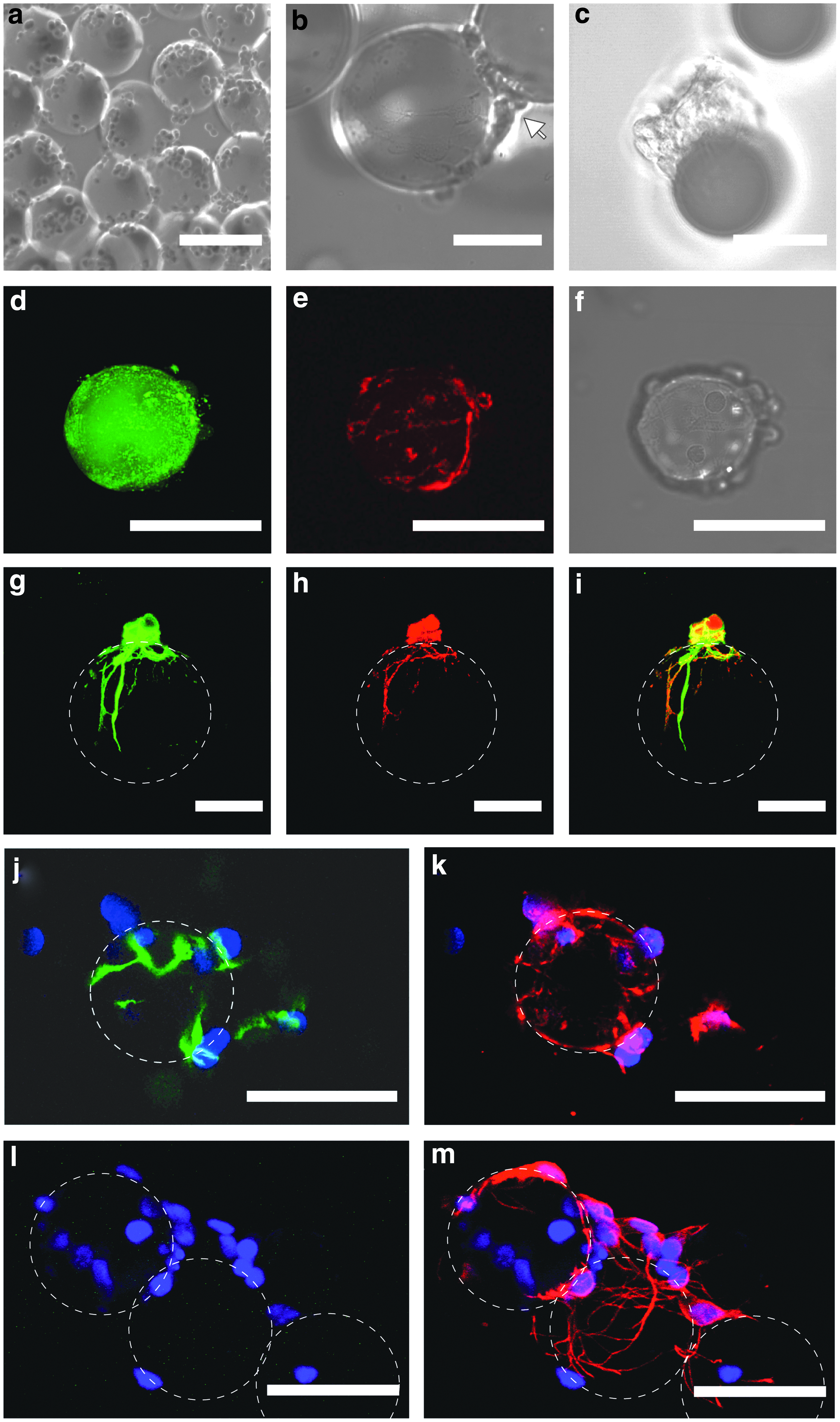
Neuronal cell maturation on MGPs
2D, two dimensional; 3D, three dimensional.
Scanning electron microscopy (SEM) at DIV 6.5 revealed differences in cell morphology between hard glass and soft hydrogel particles. Neurons grown on glass particles have developed a dense mesh of processes bridging particles (Fig. 4a–c). These processes were straight cable running between beads that were torn (arrow, Fig. 4c) rather than being peeled off from the glass particle during sample handling. On the contrary, neurons grown on MGPs exhibited processes with less directionality, suggesting that lighter MGPs might apply less constraints on neuronal processes (Fig. 4d–f). Despite the density of processes formed at the surface, neurons could be separated during sample handling with minimum damage to neuronal processes running over their surface (Fig. 4e, f). All imaged aggregates showed hollow spaces left vacant by MGPs during the thermoswitching cycle that occurred during sample preparation (Fig. 4e, open circle). After removal of the microgel carriers, higher magnification of the cell surface showed cell processes that were remarkably intact (Fig. 4f). These observations suggest that shell-coated MGPs have the dual properties to effectively promote neuronal cell adhesion and growth processes, and to allow for separation of differentiated neurons during thermoswitching without enzymatic action, thus fulfilling the prerequisite conditions to serve as cell transplant carriers.
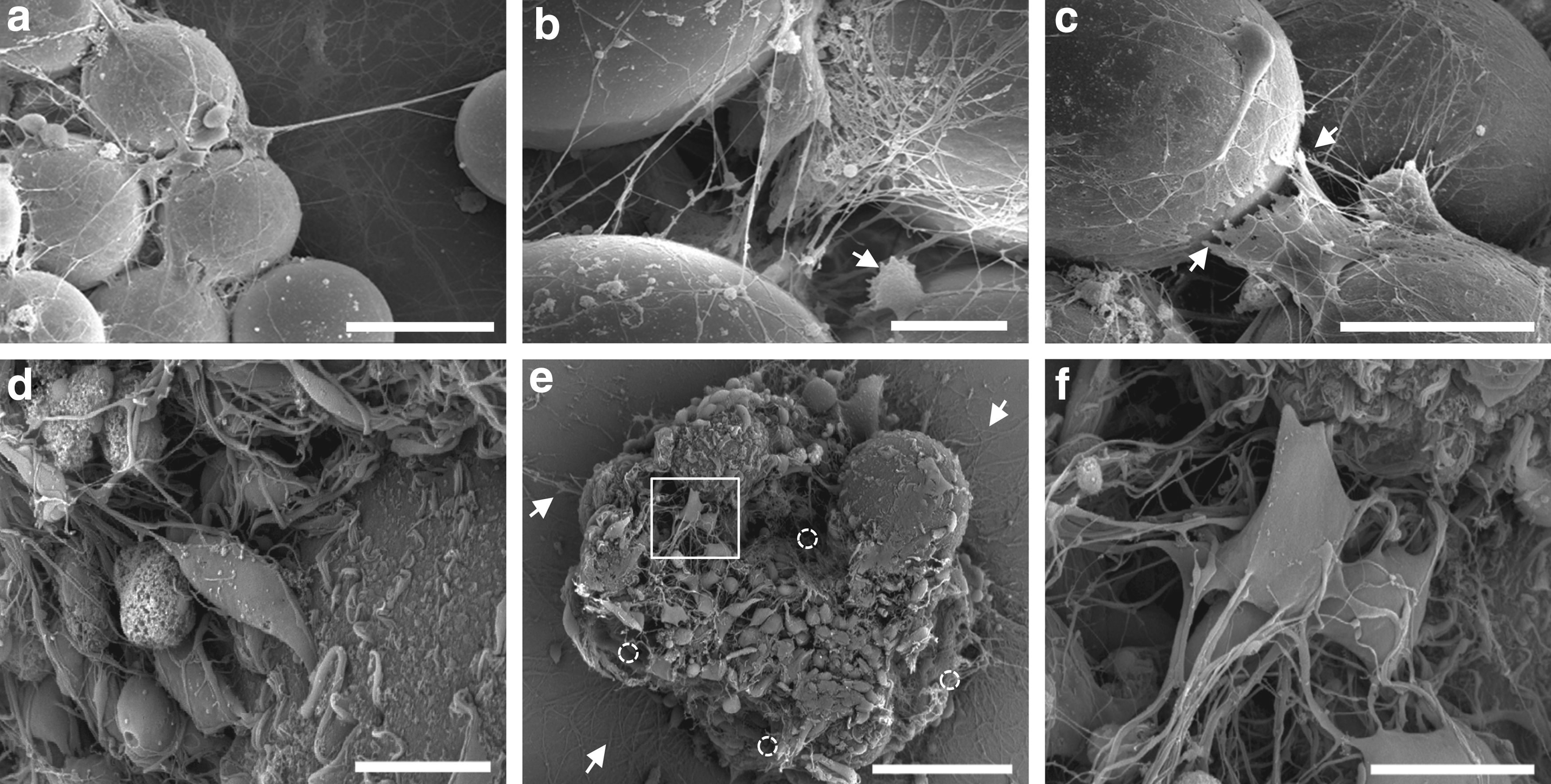
Comparison between Glass and pNIPAM
To determine the efficacy of MGPs as neuronal cell carriers compared with solid glass particles, we carried out a side-by-side transplantation study. Each carrier type was transplanted bi-laterally as described in Table 2. Particles bearing E18 hippocampus neurons labeled with a lentiviral vector driving GFP expression to label cells were stereotactically injected into the hippocampi of 6-week-old young adult recipient rats after 5 days in culture. The procedure was performed on group of
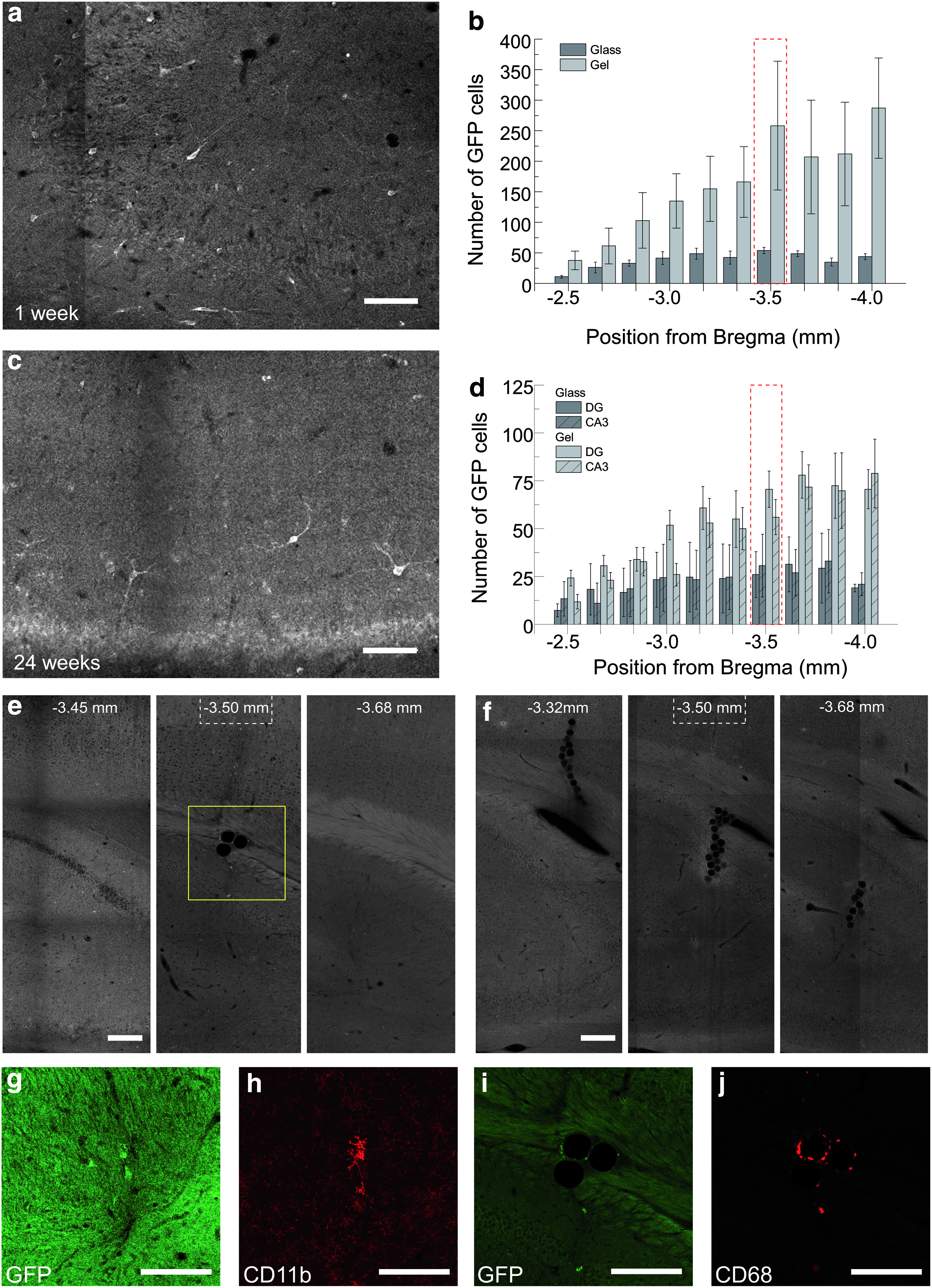
Performance of pNIPAM particles as neuronal cell transplantation carriers At 1 week post-transplantation, animals were sacrificed and their brains were sliced and immuno-stained with GFP antibody. Fluorescence microscopy images of brain slices were acquired with a laser scanning microscope.
Average of the total number of cells counted per hemisphere.
To assess cell integration as a function of the neurogenic potential of injection location, we examined the long-term maintenance of transplanted cells after their injection in either DG or Cornu Ammonis region 3 (CA3) at anterio-posterior coordinates [AP] of −3.5 mm with regard to the bregma. Coronal brain sections from animals sacrificed at 6 months post-transplantation were stained and imaged. In every section analyzed, GFP+cells with healthy morphologies were visible (Fig. 5c, for the entire coronal section see Supplementary Fig. S1), although this signal was weaker than the one observed at 1 week post-transplantation. At 6 months post-transplantation, the overall anterio-posterior cell distribution mirrored that observed after 1 week, suggesting that cell survival was independent of local cues at either the injection site or the cell's initial location. Since nonfunctionally integrated cells were typically cleared by macrophage and microglial cells, maintenance of GFP+cells suggests that at least 35% of the cells counted at 1 week post-transplantation survived and integrated with the host neuronal circuit (Table 2). Furthermore, we found that the average anterio-posterior distributions of surviving GFP+neurons between injections into CA3 versus DG were within the measurement error (Fig. 5d). We found that, regardless of the carrier type, the same percentage of short-term implanted cells remained after 24 weeks (Table 2), indicating that cells were equally likely to subsist and integrate when they were grown on microgel carriers as when they were grown on glass carriers. These results suggest that the elastic properties of the carriers did not alter the ability of cells to integrate with host cells. The most notable difference observed between treatments was the very low number of MGPs found in brain sections. At most, three MGPs were found within 250 μm anterio-posterior of the injection site in the hippocampus shown in Figure 4e, whereas glass particle aggregates were repeatedly found along the injection track as shown in Figure 4f. 25 Since pNIPAM particles were not degradable under the experimental conditions, low quantities of MGPs suggest that MGPs separated from cells during the handling procedure before injections.
To evaluate the long-term inflammation response elicited by the cell transplantation, we examined the distribution of activated macrophage and microglia cells typically associated with clearance of nonfunctionally integrated cells and foreign objects in brains at 24 weeks post-transplantation. Entire coronal brain sections taken around the injection location were immuno-stained for CD68, a macrophage marker, and for CD11b, a microglia cell marker. Confocal imaging showed only sporadic signs of macrophage and microglial activity outside the injection scar, and this activity was typically associated with the presence of MGPs (Supplementary Fig. S1). The detailed CD68 and CD11b signals around MGPs are shown in Figure 5g–j. At these sites, the GFP fluorescence signal was weak, and it could not be determined with confidence that macrophages and microglia cells were associated with nonintegrated GFP+neurons. Furthermore, only three MGPs were observed in 13 injected hippocampi, which limited our abilities to assess for a tissue inflammation response. These results suggest that both materials elicit minimal tissue response in the host.
Discussion
In this study, we evaluated the efficacy of pNIPAM polymer colloidal carriers to support the culture and neuronal cell differentiation as well as to promote cell survival post-transplantation in comparison with a previous method implementing glass particles. We demonstrate that shell-coated, soft pNIPAM particles could advantageously replace glass carriers to transplant neurons in the hippocampus. These findings demonstrate that (1) our coating protocol enables E18 hippocampal neurons to grow faithfully on thermoswitching MGPs to yield differentiated neurons that can be transplanted into young adult rats; (2) changes in the elastic properties of carriers did not change the cell composition; (3) MGPs can increase the implanted cell number by 2.7-fold compared with glass carriers after 24 weeks; (4) long-term transplanted cell survival and integration is an intrinsic property of E18 cells grown on colloidal particles, and is independent of carrier mechanical properties; and (5) cells dissociated from the MGPs during cell culture handling before transplantation minimize the injection of foreign material and make pNIPAM MGPs a promising method for neuronal cell transplantation.
We believe that the primary explanation for the increase in transplanted cell number comes from E18 neuronal cell response to MGPs mechanical properties. Indeed, we have observed that, when grown on colloidal particles, a fraction of E18 neuronal cells proliferated until we conditioned the medium to promote neuron differentiation. This proliferation was more pronounced with MGPs compared with glass particles as confirmed by DNA extraction experiments. Furthermore, the low percentage of glia cells found in these cultures after conditioning the medium suggested that proliferating cells were committed to neuron lineage. We have come to the conclusion that it is unlikely that transplanted progenitor cells continued dividing post-transplantation, because we observed no change in GFP+cell density at the neurogenic zone of the hippocampus within the first 6 months post-transplantation. Hence, the increase in transplanted neurons is most likely solely due to the increase in neuron population on the MGPs during the first few days in culture.
Furthermore, pNIPAM particles' rapid thermoswitching abilities appeared to contribute to cell release and/or cell survival. Although coated MGPs presented a suitable density of adhesion sites to promote a robust neuronal cell implantation, and extensive growth of neuronal processes, a complete thermoswitching cycle resulted in the dissociation of the cells from their carrier. The neuron morphology and the proper localization of Tuj-1 and smi-312 did not reveal differences in cytoskeleton organization. However, cell dissociation on pNIPAM particle size expansion could originate from differences in proteins responsible for sensing support elastic properties and/or due to differences in extra cellular matrix proteins deposited on the carriers. SEM pictures hinted that neuronal processes extending between two beads experienced different loads most likely due to the difference in particle weight. Sensing and adapting to these parameters could involve different adhesion molecules and force transducer elements. Further studies will be required to identify these molecules and to characterize their load-dependent response.
The preparation steps leading to the cell transplantation were carried out at RT, and the combination of the particles thermoswitching with the mechanical shear applied while loading the injection syringe completed the cell separation. Once MGPs have lost their cell load, they become extremely sticky. When injecting MGP cell suspension, the injection syringe tended to clog and required extensive ethanol washes between surgeries. Hence, we think that nonspecific binding of the MGPs to the syringe and/or needle is responsible for the limited number of particles injected. In the future, an additional step should be implemented to actively trap the MGPs before transplantation.
In conclusion, we made use of the thermosensitive properties of pNIPAM particles that make this polymer a suitable product for biomedical applications. Additional functionalities could be incorporated into this class of cell carriers to optimize cell preparation. The polymeric shell enables the diffusion of molecules, and it was previously used to engineer pH-sensitive particles for drug release. 48 Hence, future cell culture strategies could harness pNIPAM small-molecule release properties to optimize cell differentiation and growth before transplantation. Furthermore, our preparation protocol is not restricted to pNIPAM carriers. The coating protocol, in particular, which promoted neuronal cell adhesion, relies on surface charge so that adhesion can occur independent of the polymer chemistry, and it is, thus, readily transferable to other polymers that have already been tested and approved for biomedical applications.
Conclusion
In this study, we have shown that by coating pNIPAM particles with a polymeric shell, we could promote E18 neuronal cell growth and differentiation on soft MGPs. This approach fulfilled the cell replacement needs. The elastic properties of the particles promoted early E18 hippocampal cell proliferation before medium conditioning promoted neuron differentiation. The thermoswitching properties enabled the release of mature neurons from these carriers before transplantation without damaging their processes, enabling the injection of a higher number of cells with almost no MGP injected. The low number of particles injected minimized the chances for adverse tissue response. Taken together, these results suggest that thermoswitching microgel carriers are promising tools for neuronal cell transplantation. Furthermore, the flexibility of the coating protocol makes this approach applicable to a large variety of charged polymers that would be biocompatible and biodegradable thermoswitchers such as polyoxazolines. This opens the door to research involving a large library of molecules. Further investigation of the cell response to physical properties of the microgel are urged to fully understand microgel-cell interactions and to engineer particles that implement the additional advantageous properties of MGPs for their use in disease models.
Footnotes
Acknowledgments
The authors wish to thank E. Callaway for providing lentiviral DNA constructs; David Schaffer and MPI-CBG animal house for helping them establish animal surgeries; Daniel Freund and Thomas Kurth for their support with SEM sample preparation and imaging advices; and Denis Corbeil, Elly Tanaka, and Carsten Werner for their valuable comments and helpful discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft (FZ 111) and from the MedDrives-Start Program.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.