
Abstract
Successful therapy for radiation-induced salivary gland (SG) hypofunction is currently unavailable; however, tissue-specific stem cells are expected to be promising candidates for SG regeneration. Here, we present our method for the establishment of single cell-derived clonal stem cells from mouse SGs and describe their characteristics. Salivary gland-derived clonal stem cells (SGSCs) were isolated and expanded in vitro by a modified subfractionation culture method. The properties of SGSCs were examined with respect to their marker expression, gene expression, differentiation potential, and in vitro immunosuppressive activity relative to bone marrow-derived mesenchymal stem cells (BM-MSCs). SGSCs appeared to largely share the characteristics of BM-MSCs based on their marker expression, whereas they differentially expressed some genes, including AQP5, E-Cadherin, Laminin, ZO-1, and COL4. SGSCs showed the ability to differentiate into fat, bone, and cartilage cell types, as well as into α-amylase-producing and hepatocyte-like cells after appropriate induction. The in vitro immunosuppressive activity of SGSCs was found to be more potent than that of BM-MSCs. These results showed that SGSCs possess the properties of MSCs with some differential gene expression and they are salivary-specific stem cells with both epithelial and mesenchymal properties. The biological functions of SGSCs and their relevance to SG epithelial progenitor cells require further investigation.
Introduction
S
There is increasing interest in therapeutic strategies for repairing and/or restoring the damaged SG in the context of tissue engineering and regenerative medicine, and stem cell therapy has recently attracted considerable attention due to their biological potential to repair damaged tissues.4–10 Although some types of stem cells have been universally applied to treat a broad range of disorders, there is a consensus that tissue-specific stem cell treatment could be a better option for the treatment of damaged tissue. Tissue-specific stem cells are known to exist in most adult tissue and to be responsible for the regeneration of their own tissues. Considering these points, the use of SG-specific stem cells could be a good therapeutic strategy for treatment of salivary hypofunction. Putative SG stem/progenitor cells were recently isolated from rodent and human SGs and shown to differentiate into hepatic, pancreatic, and salivary epithelial cells,11–14 as well as mesenchymal cells.15,16 However, it remains unclear whether different lineage-restricted progenitor cells exist within the multipotent stem cell population or if a single stem cell population gives rise to all mesenchymal and epithelial cell types in SGs. Furthermore, the similarities and differences between SG stem/progenitor cells and other adult stem cells such as mesenchymal stem cells (MSCs) have not yet been determined. To address this matter, we attempted to isolate tissue-specific stem cells from SGs and examined their stem cell properties.
We previously reported that the subfractionation culturing method is useful for clonal isolation of subpopulations from stem cell populations in bone marrow, 17 and clonal stem cells have been shown to be beneficial and safe for transplantation-based treatment. In this study, we successfully isolated and established homogenous tissue-specific stem cells from mouse SGs and characterized them for stem cell identification. Since SG-specific stem cells were considered a type of adult stem cells, their stem cell properties were compared with MSCs, a well-known adult stem cell type. We examined stem cell properties such as self-renewal, differentiation potential, cell surface marker expression, gene expression, and immunological properties. The results presented herein will greatly improve our understanding of SG-specific stem cells and further develop cell therapy for radiation-induced salivary hypofunction.
Materials and Methods
Isolation and cultivation of salivary gland-derived clonal stem cells
All experimental procedures were conducted in accordance with the Guide for Animal Experiments of the Inha University, and the experimental protocol was approved by the Institutional Animal Ethics Committee. Six submandibular glands of 4-week-old male C3H mice were dissected and minced with a scalpel, after which the minced tissue was transferred into a dissociation buffer containing 0.2% collagenase II in the Hank balanced salt solution and incubated for 30 min at 37°C with constant shaking. The dissociated tissue solution was then filtered through 70- and 40-μm cell strainers, after which the cell suspension was centrifuged at 500 g for 5 min at 4°C. Next, the supernatant was discarded and the cell pellets were resuspended with Dulbecco's modified Eagle medium (DMEM; Gibco BRL) containing low glucose, 20% fetal bovine serum (FBS; Gibco BRL), and 1% penicillin/streptomycin (Gibco BRL) and then incubated in a 100-mm culture dish. Following incubation for 2 h at 37°C under 5% CO2, the cell culture supernatant was transferred to a new 100-mm dish and incubated for another 2 h, at which time, the supernatant was again transferred to a new dish (D1) and incubated for 2 h. The supernatant was subsequently transferred to another new dish (D2), incubated for 1 day, and transferred to another new dish (D3). This process was repeated two more times with 1- and 2-day incubations (D4 and D5, respectively). In the first two transfers, we allowed cell clumps to settle to the bottom of the dish after a short period of incubation, while longer intervals in the latter transfers were designed to allow the less dense and/or adhesive cells in the supernatant to settle to the bottom of the dish. We hypothesized that this method might allow cells with different densities and/or adherence to be further fractionated. The single-cell derived colonies that appeared in the D2, D3, D4, and D5 dishes were detached and isolated by 3 min of treatment with 0.25% trypsin/ethylenediaminetetraaceticacid (EDTA) in cloning cylinders (Gibco BRL), after which they were transferred to a six-well plate and then to larger culture flasks, where they continued to expand (Fig. 1). For comparison with other adult stem cells, bone marrow-derived mesenchymal stem cells (BM-MSCs) were obtained as previously described. 18

Subfractionation culturing method to obtain single cell-derived, highly homogenous clonal stem cells from salivary glands (SGs). Color images available online at
Cell proliferation and colony-forming unit assays
To analyze the proliferation ability, the established SG-derived clonal cells or BM-MSCs were seeded at a density of 1×104 per 100-mm dish in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cell counting data from passage numbers 11 to 24 were used to evaluate proliferation. During analysis, the culture medium was replaced every 2 days and the cells were counted every 3 days. For colony-forming unit (CFU) assay, salivary gland-derived clonal stem cells (SGSCs) and BM-MSCs at passage 5 were plated at a density of 100 cells per well in a six-well plate in triplicate. The cells were then cultured for 10 days, and the medium was changed every 3 days. At the end of the culture period, cells were fixed with 10% formalin for 1 h and then stained with 0.5% crystal violet for 10 min. All colonies positive for crystal violet were counted. The CFU activity was expressed as percent formation, which was calculated as follows: average number of colonies formed/total number of seeded cells×100. Three independent experiments were conducted for the CFU assay.
Flow cytometric analysis for cell surface maker expression
Briefly, established SGSCs at passage 9 and cultured BM-MSCs at passage 16 were subjected to flow cytometry for analysis of 12-cell surface marker proteins. The cells were washed twice with phosphate-buffered saline (PBS) and then harvested by treatment with trypsin/EDTA. Next, cells were incubated with the fluorescein isothiocyanate or phycoerythrin-conjugated antibodies, after which they were analyzed using the FACSCalibur system (BD Biosciences), and data were analyzed using the CellQuest software (BD Biosciences). The antibodies used for flow cytometric analysis were as follows: CD44 (Serotec), Sca-1 (BD Biosciences), CD29 (Biolegend), CD49f (R&D Systems), CD34 (Serotec), CD45 (BD Biosciences), CD73 (e-Biosciences), CD90 (R&D Systems), c-Kit (BD Biosciences), AQP5 (Bioss), MHC-class I (BD Biosciences), and MHC-class II (BD Biosciences). Isotype-matched control antibody was used in each antibody analysis.
Gene expression analysis
Total RNA was extracted from SGSCs at passage 6, BM-MSCs at passage 5, and SG primary cells from C3H submandibular glands served as the control group. cDNA was synthesized from the total RNA (1 μg) using a reverse transcription system kit (Bioneer) according to the manufacturer's instructions. Polymerase chain reaction (PCR) was then carried out using primers specific for each gene (Table 1). PCR was conducted by subjecting the samples to 30–40 cycles of denaturation at 94°C for 30 s, annealing at 53–60°C for 30 s, and extension at 72°C for 30 s, after which the amplified DNA products were run on a 1.5% agarose gel.
Immunofluorescence staining
Cultured cells were washed two times with PBS, fixed with methanol at −20°C for 10 min, and then washed in PBS containing 0.05% Tween 20. Nonspecific binding was blocked with PBS containing 1% bovine serum albumin. The cells were then incubated with primary antibody for AQP5 (Calbiochem), c-Kit (CD117; Santa Cruz Biotechnology), Ascl-3 (Santa Cruz Biotechnology), CK-5 (Abcam), and collagen IV (EMD Millipore) in a moist chamber overnight at 4°C. After washing in PBS, cells were incubated with Alexa-488-conjugated goat anti-rabbit immunoglobulin G for 2 h in the dark at room temperature. Next, 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; Vector Labs, H-1500) was added for 3–5 min to stain the cell nuclei. All experiments included a slide with no primary antibody as a negative control. After mounting, cells were viewed using a confocal laser scanning microscope (Olympus FV1000; Olympus).
In vitro differentiation
SGSCs were analyzed for their capacity to differentiate into mesenchymal cells (adipogenic, osteogenic, and chondrogenic), as previously described. 18 SGSCs were also examined to determine whether they could differentiate into salivary epithelial and hepatic cell lineages. For SG epithelial differentiation, cells at passage 6 were seeded at 1×104 cells per well onto plates that had been precoated with Matrigel (BD Biosciences). NIH3T3 normal fibroblasts served as a control. The cells were then cultured in DMEM/F12 (Gibco BRL) supplemented with 10% FBS, 1% penicillin/streptomycin, 1% ITS premix, 10 mM nicotinamide (Sigma), 1×10−7 M dexamethasone (Sigma), 1 mM β-mercaptoethanol (Gibco BRL), 20 ng/mL epidermal growth factor, and 20 ng/mL hepatocyte growth factor (R&D Systems) at 37°C for 3 days and then photographed under a light microscope (Olympus CKX41; Olympus). Next, the cells were used for RNA analysis and immunofluorescence staining. Anti-a-amylase antibody (Cell Signaling Technology) was used for immunofluorescence staining.
For hepatogenic differentiation, the cells were seeded at 1.5×104 cells/cm2 onto Matrigel-coated surfaces and allowed to adhere to the surfaces. Adherent cells were subsequently incubated in the hepatogenic basal medium (DMEM/F12 supplemented with 1% glutamine [Gibco BRL] and 1% nonessential amino acids [Gibco BRL]) without serum for 1 day. The starved cells were subsequently cultured in the hepatogenic basal medium containing 100 ng/mL activin (R&D Systems) and 10 ng/mL basic fibroblast growth factor (bFGF; R&D Systems) for 3 days, after which they were incubated in the basal medium supplemented with 20 ng/mL hepatocyte growth factor (R&D Systems), 10 ng/mL FGF4 (R&D Systems), 0.61 g/L nicotinamide (Tocris), 10 ng/mL bFGF, and 0.1 mM ascorbic acid (Sigma) for an additional 7 days. The next step was accomplished by incubation in the presence of 20 ng/mL oncostatin M (Gibco BRL), 20 ng/mL hepatocyte growth factor, 10−7 M dexamethasone, 0.1 mM ascorbic acid, and 1% ITS premix for 3 days. The cells were then cultured in the basal medium containing 20 ng/mL oncostatin M, 10−7 M dexamethasone, 0.1 mM ascorbic acid, 1% ITS premix, and 1% FBS for 21 days. During the experiment, the medium was changed every 3 days. At the end of differentiation, cells were stained with periodic acid–Schiff (PAS) to detect polysaccharides, such as glycogen.
Each differentiation was further confirmed by mRNA expression of differentiation-specific marker genes as listed in Table 1.
In vitro immunosuppression assay
To examine the immunosuppressive activity of SGSCs against T-cell proliferation, the SGSCs were cocultured with mouse splenocytes under the conditions of antibody stimulation or mixed lymphocyte reaction (MLR) and then compared with BM-MSCs as well-established immunosuppressive cells. Splenocytes isolated from female Balb/c mice were seeded in triplicate at a density of 2×105 cells per well in a 96-well plate with or without stem cells (SGSCs or BM-MSCs) in the presence of anti-CD3 (1 μg/mL) and anti-CD28 (1 μg/mL) antibodies. The stem cells were then cocultured with fixed numbers of splenocytes at ratios of 1:20, 1:50, and 1:100 (stem cells:splenocytes) for 48 h. For MLR, two different splenocyte populations were isolated from female Balb/c and C57BL/6. Next, 1×105 cells from each population were cocultured together with stem cells (SGSCs or BM-MSCs) at ratios of 1:20, 1:50, and 1:100 (stem cells-to-splenocytes ratio) for 96 h. Following coculture of the splenocytes and the indicated stem cells under both experimental conditions, the proliferation of splenocytes was determined by [ 3 H]-thymidine incorporation for an additional 16 h. Finally, the radioactivity was measured in the MicroBeta2 counter (Perkin Elmer).
Results
Isolation of clonal stem cells from SGs
We attempted to isolate tissue-resident adult stem cells from mouse SGs using a method based on subfractionation culture that has been demonstrated to be very effective for isolation of highly homogenous clonal MSC from BM (Fig. 1). 17 We obtained a number of single cell-derived colonies from submandibular glands of male C3H and then cloned the plastic-adherent colony-forming cell populations. To establish clonal SG-derived stem cell populations, several clones of single colony-derived SG cells were subject to large-scale expansion followed by cell characterization. We selected clone-17 as a putative SG-specific adult stem cell population and examined its biological properties, considering the cell growth, morphological consistency, and colony formation activity after subculture.
Self-renewal and CFU activity
Upon light microscopy, clonal cells cultured on plastic tissue plates showed a fibroblast-like appearance. During subculture, morphological consistency was maintained in a monolayer culture (Fig. 2a). The self-renewal properties of stem cells were evaluated by long-term proliferation and CFU activity. The cells constantly proliferated during long-term cultivation (up to passage number 24), indicating that the isolated clonal cells are highly proliferating rather than dormant or quiescent (Fig. 2b). To measure CFU efficiency, SG cells or BM-MSCs at passage 5 were seeded at 100 cells per well in a six-well plate and then cultured for 10 days, after which the cells were stained with crystal violet for apparent visualization. All colonies formed were counted, and the CFU activity was calculated as described in the Materials and Methods section. As shown in Figure 2c, more colonies were formed in the SG cells than in BM-MSCs. The CFU efficiency of the SG cells (64.0±5.0%) appeared to be higher than that of BM-MSCs (35.7±2.9%), indicating that the isolated clonal SG cells have stem cell properties of self-renewal.

Morphology and proliferation of clonal salivary gland-derived cells (SGCs) following passages in the subculture.
Stem cell marker analysis
We compared the clonal SG cells with MSCs to determine if the SG cells exhibited characteristics typical of adult stem cells. To examine whether the isolated SG cells possess MSC-like properties, we performed flow cytometric analysis of MSC-specific cell surface marker expression. For comparison, MSCs isolated from BM of the same mouse strain were analyzed in parallel. Similar to BM-MSCs, SG cells were positive for CD29, CD 44, CD73, CD49f, Sca-1, and MHC class I, but negative for CD34, CD45, CD90, c-Kit, or MHC class II (Fig. 3a). Notably, differential expression of AQP5 was observed between SG cells and BM-MSCs. AQP5 was expressed in the clonal SG cells, but not in BM-MSCs (Fig. 3a). After repeated subcultivation of the cells up to passage number 13, AQP5 expression in the clonal SG cells was stably maintained (Fig. 3b).

Comparison of cell surface marker expression between the isolated SGCs and BM-MSCs.
To further examine the tissue-specific differential gene expression, basal mRNA expression of some SG-specific genes between the SG cells and BM-MSCs was compared (Fig. 4a). SG tissue was included as a positive control. As expected, AQP-5 mRNA was detected in both the SG cells and the gland tissue, but not in BM-MSCs. AQP5, which encodes a water channel protein, aquaporin-5, plays an important role in the generation of saliva. The SG cells highly expressed some epithelial genes, including E-Cadherin, Laminin, and ZO-1 (Fig. 4a), whereas BM-MSCs did not. COL4 encoding type IV collagen was also highly expressed in the SG cells but weakly expressed in BM-MSCs (Fig. 4a). Type IV collagen is a major component of the basement membrane, which is essential for salivary epithelium. However, basal expression of AMY encoding a functional salivary marker (α-amylase) was not observed in culture-expanded SG cells or BM-MSCs (Fig. 4a). ASCL3 encoding a transcription factor achaete–scute complex homolog 3 (Ascl3) has recently been suggested as an adult progenitor cell marker of mouse SGs.19,20 Basal mRNA expression of ASCL3 was not observed in SG cells or BM-MSCs (Fig. 4a). Furthermore, we did not observe expression of CK5 encoding cytokeratin 5 in either cell. Moreover, no differential gene expression of NKCC1 (for Na-K-2Cl cotransporter), CK19 (for cytokeratin 19), and COL1 (for type I collagen) was observed between SG cells and BM-MSCs. Among these genes, we evaluated the protein expression of three genes, including AQP5, COL4, and E-Cadherin. Consistent with the gene expression data, differential protein expression of AQP-5 and type IV collagen between SG cells and BM-MSCs were observed by IF staining (Fig. 4b). Exclusive expression of E-Cadherin in only the SG cells was confirmed by western blot analysis (Fig. 4c).
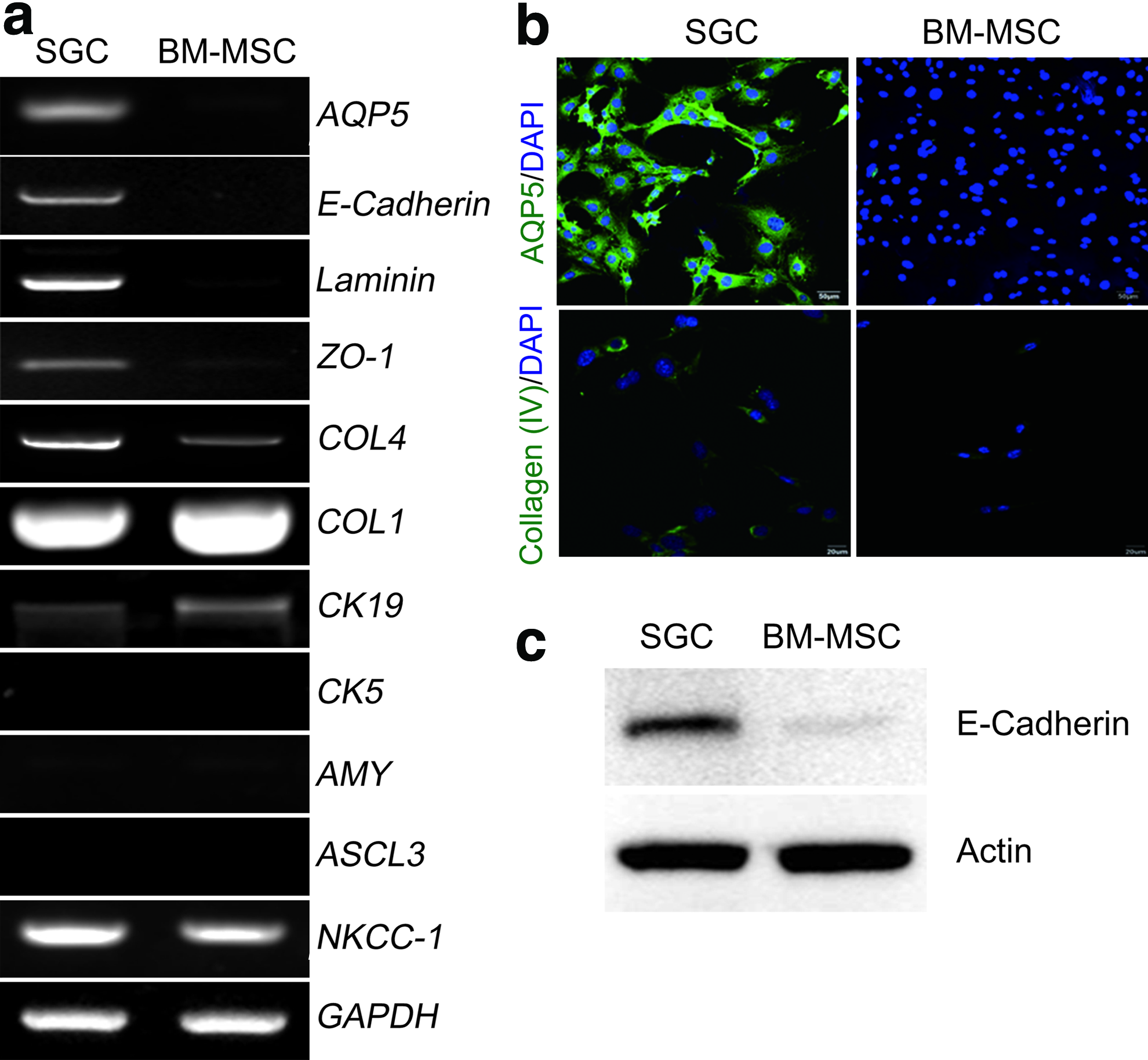
Differential gene expression between SGCs and BM-MSCs.
Differentiation potential
We next explored whether SG cells possess the multipotency to differentiate into various cell types. Evaluation of the mesenchymal differentiation potential revealed that the SG-derived clonal cells successfully exhibited fat, bone, and cartilage phenotypes upon cultivation on adipogenic, osteogenic, and chondrogenic media, respectively (Fig. 5a). Lineage-specific molecular markers were concomitantly expressed during each differentiation. Specifically, LPL, FABP4, and PPARγ2 were representative adipogenic markers, ALP, RUNX2, and DLX5 were osteogenic markers, and ACAN (for aggrecan) and COL10 (for type X collagen) were chondrogenic markers (Fig. 5b). 18
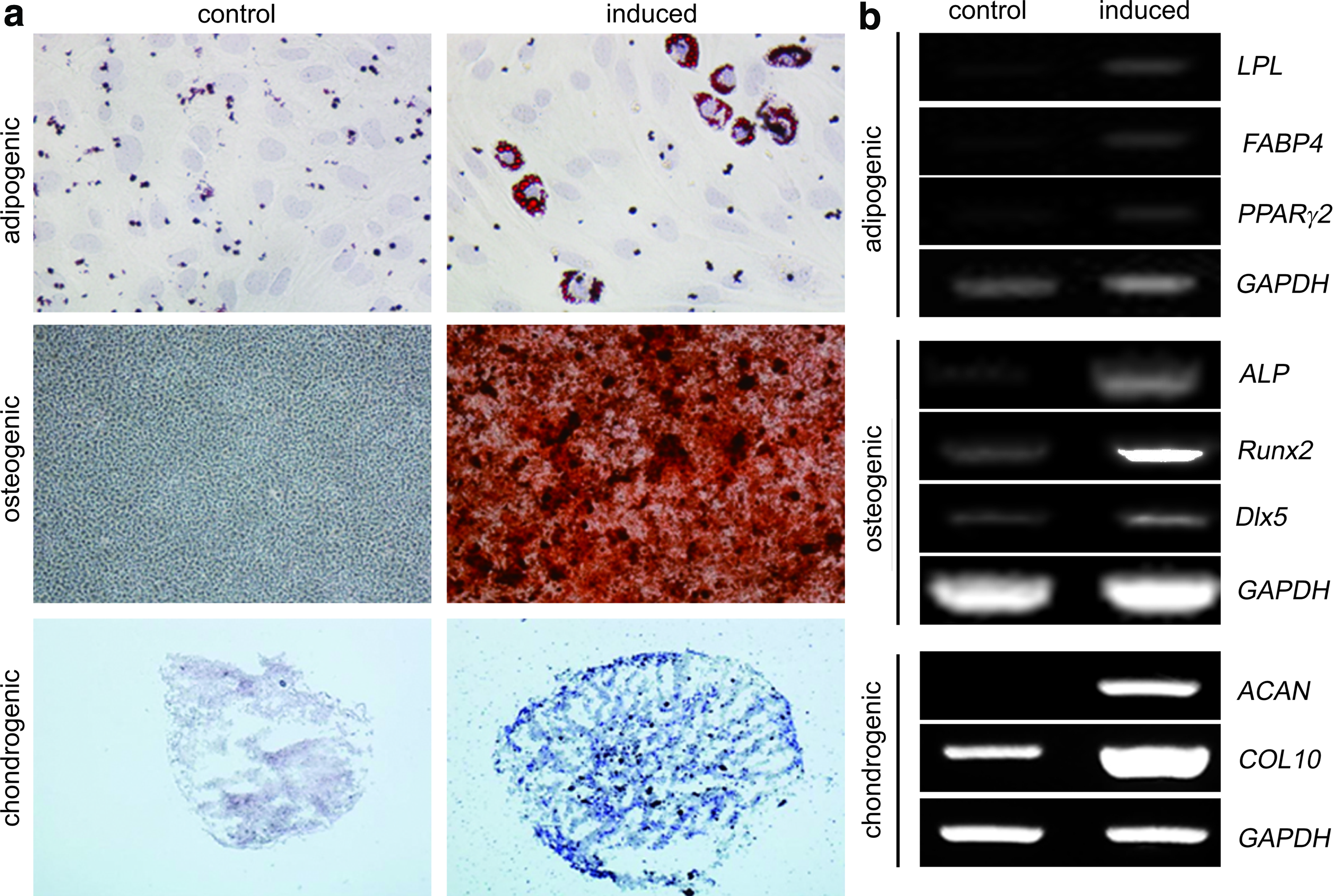
The multipotent differentiation capabilities of salivary gland-derived clonal stem cells.
To evaluate the epithelial differentiation potential of the SG cells, we assessed whether SG cells differentiate in vitro into salivary epithelial cells and hepatocytes. SG cells seeded on Matrigel-coated matrix showed morphological changes within 3 days of an appropriate induction (Fig. 6a). Specifically, at 3 days postinduction, the induced cells expressed α-amylase (Fig. 6b). However, α-amylase induction was not observed in NIH-3T3 fibroblasts cultivated in the same induction medium, implying that the SG cells possess the potency to differentiate into salivary acinar cells. Salivary acinar cell differentiation of the SG cells was further supported by induction of AMY mRNA expression (Fig. 6c). In addition, SG-derived clonal cells exhibited hepatocyte phenotype upon hepatogenic induction. Hepatogenically differentiated cells were positive for PAS staining, which is specific to glycogen in liver hepatocytes (Fig. 6d). Hepatocyte-specific marker genes, such as Albumin, α-fetoprotein (AFP), α1-anti-trypsin (AAT), tyrosine aminotransferase (TAT), and Annexin I, were significantly upregulated during late-stage differentiation (Fig. 6e). Collectively, these results indicate that SG-derived clonal cells have epithelial multipotency as well as mesenchymal differentiation potential.

Differentiation of SG cells into salivary epithelial cells and hepatocytes.
In vitro immunosuppressive activity
One of the characteristics of MSCs is their inherent immunomodulatory or immunosuppressive activity. Therefore, we examined whether the isolated SG-derived clonal cells possess immunosuppressive activity as MSCs by a [ 3 H]-thymidine incorporation assay. Under the conditions of splenocyte stimulation induced by anti-CD3/CD28 antibodies, coculture of the SG cells significantly inhibited the proliferation of splenocytes in a cell number-dependent manner, similar to BM-MSCs (Fig. 7a). The SG cells also dramatically inhibited the proliferation of splenocytes in a mixed lymphocyte reaction (Fig. 7b). In these assays, the in vitro immunosuppressive activity of the SG cells appeared to be more potent than that of BM-MSCs.
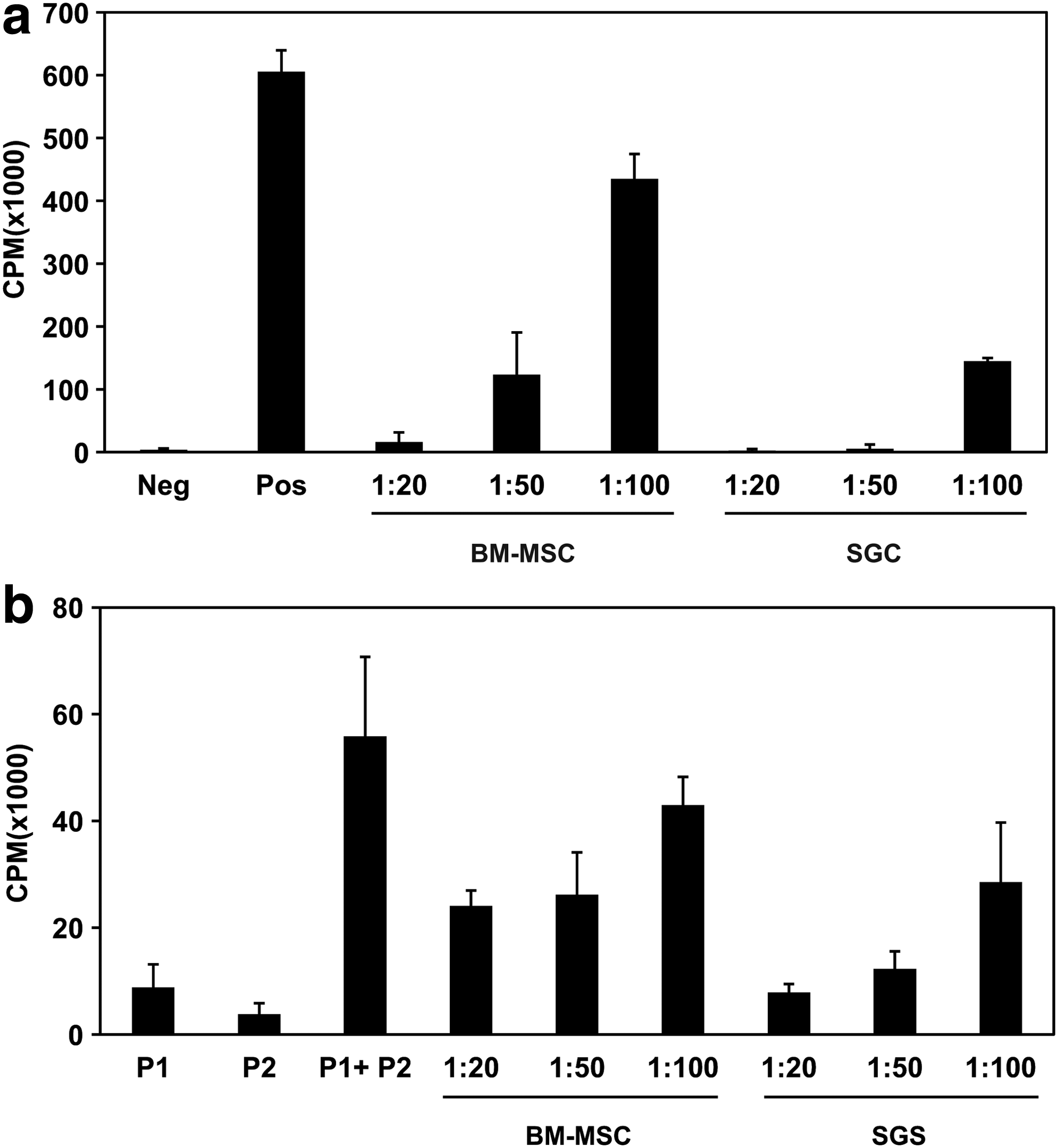
Immunomodulation activity of SG cells and BM-MSCs by in vitro immunosuppression assays.
Discussion
In this study, we successfully established clonal SG cells by a subfractionation culture based method. Our data provide evidence that the isolated clonal SG cells may be a novel type of tissue-resident SG stem cells (SGSCs) that share MSC properties but have distinct potential for transdifferentiation into salivary epithelial cells. SGSCs and BM-MSCs were found to have similar marker expression patterns, differentiation potential, and in vitro immunosuppressive activity. Although our data unambiguously indicate that the isolated clonal SGSCs possess properties typical of MSCs, there were discrepancies between SGSCs and BM-MSCs. For example, some epithelial-related genes, such as AQP5, E-cadherin, Laminin, ZO-1, and COL4, were differentially expressed between these cell types.
Production of highly homogeneous SGSCs is a critical consideration for clinical applications. Although conventional methods, such as selective low-density culture systems13,16 and floating cell culture, 21 have been used in attempts to isolate and expand SGSCs, it is not clear how many types of stem/progenitor cells are heterogeneously mixed in culture-expanded cell products. Moreover, the isolated putative stem cells readily undergo cell death or differentiation during ex vivo expansion due to inappropriate isolation and culture methods. Therefore, well-established stem cell isolation protocols and subsequent cell expansion processes for high-quality stem cell maintenance are required for preparation of SGSCs.
Researchers have attempted to isolate SG stem/progenitor cells from SG-derived cells by cell sorting using cell surface markers, including c-Kit, 5 Sca-1, 12 and α6β111,22 in rodents and CD49f (integrin α6), Thy-1, 14 and c-Kit 21 in humans. However, since flow cytometric analysis revealed that only a few cells (0.065% of c-Kit expressing cells from salispheres) constitute the population, 23 this method appears to be inefficient for obtaining a number of SG progenitor cells from patients. There has been another approach for isolation of SG progenitor cells after stem cell enrichment through salivary duct ligation-induced atrophy or heat stress.11,12,22 Nevertheless, this method may not be an alternative because it is difficult to employ these conditions for clinical applications.
We attempted to identify another method to isolate and obtain SGSCs using a subfractionation culturing method that has been demonstrated to be efficient for highly homogeneous clonal MSC isolation and expansion. 17 When compared with the conventional MSC isolation method, this method is relatively simple and has some advantages, such as minimized loss of source material. In this study, we successfully obtained single cell-derived clones through our method. Importantly, our method generates highly homogeneous clonal stem cells, whereas other conventional isolation methods produce mixed cell populations.
We characterized our clonal SGSCs and found that they possess MSC properties but have some characteristics distinct from typical BM-MSCs. Our data showed that SGSCs express typical MSC markers, such as CD29, CD44, CD49f, CD73, MHC-class I, and Sca-1, and that they have mesenchymal multipotency toward fat, bone, or cartilage cell types following adipogenic, osteogenic, or chondrogenic induction, respectively. Furthermore, the SGSCs investigated in this study exhibited intense type I collagen expression and lacked suggested markers for SG progenitor cells, such as c-Kit, Ascl-3, and CK5,19,21,24 indicating that they may be a previously unreported type of SG-derived stem cells that differ from SG progenitor cells. We compared the SGSCs investigated in this study to BM-MSCs to identify the properties of SGSCs that differed from MSCs. The results revealed that SGSCs exhibit MSC phenotypes, including multipotency to differentiate into mesenchymal cell types, in vitro immunomodulatory capacity, and a series of MSC markers. Nevertheless, there was an obvious distinction between our SGSCs and typical MSCs. Comparative examination of gene expression provided evidence that AQP5, E-cadherin, Laminin, ZO-1, and COL4 are preferentially expressed in SGSCs, but not in MSCs (Fig. 4). These findings suggest that these genes may be used to discriminate SGSCs from typical MSCs. Furthermore, the SGSCs showed the potential for epithelial differentiation (Fig. 6).
To date, several putative salivary stem/progenitor cell populations have been reported (Table 2). SG tissues contain epithelial (acinar and ductal cells) and stromal cells (myoepithelial cells, myofibroblasts, immune cells, endothelial cells, and neural cells) with different developmental cell lineages. Recent studies reported the existence of MSC-like cells expressing markers that have been suggested to be involved in mesenchymal regeneration, such as CD29, CD44, and CD105.15,16 c-Kit-positive cells were suggested to be SG progenitors uniquely isolated from salispheres. 21 The c-Kit-positive cells were only detected in excretory ducts and developed into ductal and acinar-like cells when placed in three-dimensional culture. 21 Ascl319 and CK524 have also been proposed to define salivary progenitors in experiments tracing genetic lineage. Ascl3-positive cells remained in the ducts during lineage tracing and primarily yielded ductal cells and small numbers of acinar cells, implying that these cells may be SG progenitors. CK5-expressing cells were proposed as SG basal cells with progenitor potential to differentiate to acinar and ductal cells. However, further evidence is needed to verify these findings because neither K5 nor Ascl is considered as an exclusive and unique SG progenitor marker.
MSC markers: CD29, CD44, CD90, and CD105.
MSC, mesenchymal stem cell; FACS, fluorescent-activated cell sorting; MACS, magnetic affinity cell sorting; SG, salivary gland.
In our study, SGSCs did not express the previously suggested SG progenitor markers, c-Kit, Ascl-3, and CK5, implying that they may be different from progenitor cells originated from SG ductal cells. Okumura and coworkers recently suggested that laminin-producing basal cell-like cells reside in the periductal area in contact with the basement membrane and showed that they are able to transdifferentiate into hepatocytes, pancreatic β-cells, and amylase-producing cells.14,25 Since the SGSCs used in the present study were shown to exhibit phenotypes similar to MSCs and basal-like cells, it is believed that there are tissue-resident multipotent stem cells possessing mesenchymal and epithelial properties in salivary tissue. In this context, SG homeostasis and remodeling may involve coordinated interaction of more than a single type of stem/progenitor cells since the SGSCs investigated herein are a different type of cells from the suggested SG progenitor cells. Further investigation is needed to determine whether MSC-like stem cells and periductal basal cell-like progenitor cells in SG tissues are subsets of the same adult stem cell type or are different stem cell populations with distinct biological functions.
SG stem/progenitor cells have been investigated for their potential therapeutic application to ameliorate and regenerate irradiation-afflicted SGs following radiotherapy for the treatment of patients with head and neck cancers. In principle, biopsy samples should be taken from the patient's healthy SG before radiotherapy to isolate, expand, and store highly homogeneous stem cells until the end of therapy to ensure functional SG tissue regeneration. The key strategy in SG regeneration is to isolate and obtain sufficient numbers of stem cells with effective differentiation potential into specific cell types. Lombaert et al. found that intraglandular transplantation of c-Kit-positive SG progenitor cells could restore SG tissue integrity and function following radiation damage. 5 Here, we report that SGSCs are readily isolated from SGs and can be expanded ex vivo for many generations by our protocol. In addition, we provide evidence that the SGSCs investigated herein can suppress the proliferation of lymphocytes as potent as BM-MSCs. The remarkable immunosuppressive activity of SGSCs indicates that they may be a promising cell therapeutic option to improve SG hypofunction by reducing inflammation induced by radiation-inflicted SG damage.
In conclusion, our results showed that (1) the SGSCs expanded ex vivo by the subfractionation culturing method in the current study shared MSC properties in terms of proliferation, mesenchymal differentiation potential, stem cell marker expression, and immunosuppression. (2) SGSCs differentially expressed epithelial-related markers of AQP5, E-cadherin, Laminin, ZO-1, and COL4 and could differentiate into amylase-producing cells and hepatocytes. (3) These SGSCs did not express markers for SG progenitor cells, such as c-Kit, Ascl-3, and K5.
Taken together, although SGSCs share MSC-like properties with regard to proliferation, multilineage differentiation potential, and immunomodulatory capacity, the SGSCs are salivary-specific stem cells with both epithelial and mesenchymal properties. Our observations also suggest that there are likely MSC-like salivary-specific stem cells, which are different from SG progenitor cells. Although further investigation is required to unveil the in vivo location and biological roles of SGSCs in development, repair, and regeneration, we speculate that SG homeostasis and remodeling may involve coordinated interaction of more than a single type of stem/progenitor cells, and the SGSCs investigated in this study may participate in salivary epithelial regeneration.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program maintained by the Korean Society of Head and Neck Surgery and by an Inha University Research Grant. The funders played no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Disclosure Statement
No competing financial interests exist.