
Abstract
In this article, we propose a systemic approach to investigate the impact of electrohydrodynamic jetting (EHDJ) encapsulation on viability, proliferation, and functionality of the encapsulated cells. EHDJ consists in applying a high-voltage electrical field between a target substrate and a jetting needle, which is fed with a suspension of cells in a polymeric solution undergoing a sol–gel transition upon contact with the target. The viability, proliferation, and self-assembling ability of SHSY5Y human neuroblastoma cell line encapsulated in 2% alginate microbeads were analyzed by confocal microscopy and DNA quantification assays. In addition, the expression of stress (HSP70B′), apoptotic (CASP3), necrotic (HMGB1), hypoxic (HYOU1, GAPDH), and adhesion (CDH2) markers was measured with reverse transcription quantitative polymerase chain reaction (qPCR). After an initial upregulation of the HSP70B′ expression within 24 h, its expression decreased to the negative control level together with a decrease in the expression of CASP3. Any increase in necrotic or hypoxic marker expression was not detected, while a slight upregulation of CDH2 was observed in the first days after encapsulation, followed by its downregulation and stabilization to the control level. Furthermore, cell-laden beads started to self-assemble in three-dimensional (3D) constructs from the 3rd week after encapsulation. The results indicated that the EHDJ encapsulation method had a mild effect on cells, which after a week, fully recovered their proliferation rate and ability to self-assemble into 3D constructs.
Introduction
C
In regenerative medicine, cell encapsulation has received attention for the reconstruction of tissues or organs, following an early work of Mironov 7 who introduced the concept of organ printing. The procedure consists in the layer-by-layer deposition of encapsulated cells that are assembled in three-dimensional (3D) structures8,9 to reproduce the organ/tissue architecture. Three-dimensional cell-laden structures may also be suitable models for the fundamental science, drug screening, as well as a platform for diseases diagnostics.7–11
Many methods have been proposed for cell encapsulation, such as microfluidics, emulsion, stereo/photolithography, and extrusion. 1 These methods require fine tuning of several parameters and an accurate selection of the encapsulating materials to keep cells alive, active, and capable of carrying out their main functions, such as the ability to self-assemble 12 or to produce biomolecules. 13 In the previous years, cell encapsulation has been proposed for applications in different fields, like bioprinting or skeletal tissue engineering.14–16 Agarose, gelatin, chitosan, collagen, polyethylene glycol, hyaluronic acid, alginate, and other materials have been considered for the encapsulation of cells.14–16
Viability/activity of encapsulated cells has already been investigated.1,17–19 Examples are the evaluation of the heat shock protein (HSP) 60/70 expression in bovine aortic endothelial cells printed by laser pulses 1 ; the analysis of gene expression on some housekeeping, nonspecific, and blood-specific genes in bio-electrospraying assay of whole human blood 18 ; the determination of the immune response on the presence of allogeneic/xenogeneic hepatoma cells encapsulated in the beads and transplanted in rats. 19
One still open issue is whether encapsulated cells keep their original functionality, gene expression profile, and ability to self-organize in tissues/organs.
In this study, cells were encapsulated in alginate by the electrohydrodynamic jetting (EHDJ) technology.1,13,20
Alginate is a natural polymer derived from the cell wall of brown algae, which quickly crosslinks upon contact with calcium-containing solutions and can dissolve in vivo due to the calcium substitution.2,14 Behavioral parameters of SHSY5Y neuroblastoma cell line, selected as a model, encapsulated by EHDJ in alginate microbeads were assessed in short term from 3 h up to 7 days and in the long term, up to 4 weeks. Cell viability, activity, and proliferation, as well as ability of cells to self-assemble, and gene expression of stress state-related markers (CASP3, HMGB1, HYOU1, and HSP70B′) were evaluated in this study. Upregulation of CASP3 (caspase-3) gene—a late apoptosis executor, indicates the induction of apoptosis in cells21–24 ; HMGB1 (high mobility group box protein 1) is a marker that is specifically produced in necrotic conditions 21 ; HYOU1 (HYpOxia-Upregulated 1) is a hypoxia inducible gene25,26; and the housekeeping gene GAPDH (glyceraldehyde-3-phosphate dehydrogenase) is also overexpressed in hypoxic conditions.27,28 HSP70B′ (heat shock protein 70B′) is specifically produced by cells in stress conditions.29–31 Finally, cell adhesion, motion, and communication were evaluated by NCDH (N-CaDHerin) expression.32,33 The expression of all the above genes in the samples was compared to the negative control (cells grown in a tissue culture plate [TCP] till 85–90% confluence) and to the positive control (with induced hypoxia, apoptosis, necrosis, and heat shock).34–36 The gene expression level was normalized to the housekeeping control gene RPS18 (40S ribosomal protein S18) expression. 37
This article, therefore, presents a systemic investigation of the influence of EHDJ encapsulation on cells, with reference to the duration of encapsulation and cell viability, their ability to self-assemble, proliferate, and the expression of stress-related and some other vitally important genes.
Materials and Methods
Materials
Alginic acid sodium salt from brown algae (alginate) and calcium chloride dehydrate were purchased from Sigma-Aldrich. Calcein-AM, propidium iodide, phosphate buffer saline without calcium and magnesium (PBS), and Dulbecco's modified Eagle's medium (DMEM) were purchased from Invitrogen. The encapsulation system consists of a generator (ES30; Gamma High Voltage Research, Inc.), a pump (NE-300; New Era Pump Systems), a polytetrafluoroethylene tube, and a 33-gauge stainless steel needle (outer diameter: 0.210 mm, inner diameter: 0.108 mm; Hamilton). The SHSY5Y human neuroblastoma cell line (ATCC® CRL-2266™), Sonicator (UP400S; Heilscher), Quant-iT PicoGreen dsDNA Assay Kit (catalog number: P11496; Invitrogen), 0.05% Triton-X 100 in PBS, and the RNeasy Plus Mini Kit (catalog number: 74134; QIAGEN) have been used
Encapsulation process
Hydrogel preparation and sterilization
Alginate powder was dissolved in PBS for 8 h at room temperature under mild stirring to obtain an alginate solution at the desired concentration (2%, 2 g/100 mL). Under a biological sterile hood, a syringe filled with solution was connected to a pump to be filtered overnight through a 0.22-μm filter. The sterile solution was immediately used after filtration. The crosslinking solution consisted of calcium chloride dehydrate dissolved in distilled water at a concentration of 400 mM and filtered through a 0.22-μm filter under sterile conditions.
Cell culture
The SHSY5Y human neuroblastoma cell line was expanded in 25–175-mm tissue-treated culture flasks as monolayer (passages 12–16) at 37°C under 5% CO2 in high-glucose DMEM with 10% fetal bovine serum (GIBCO), 2 mM glutamine, and 1% penicillin–streptomycin mixture (Sigma). DMEM was changed every third day. The cells were cultured to 90–95% confluence before encapsulation. 38
Preparation of the alginate suspension with cells
At 90–95% confluence, cells were detached and moved to a 15-mL vial. Cells inside the vial were stirred at 1000 rounds/min for 10 min and the supernatant was removed. Cells on the bottom of the vial were resuspended in PBS and stirred again to remove any residues of medium containing cations that could crosslink the alginate solution. Cells were dispersed by vibration inside the buffer and an aliquot of the solution was taken to count the number of cells using a Cellometer Auto T4 (Nexcelom) and Trypan Blue 0.4% (Life technologies) as contrast agent. Cells were stirred again, and after removing the supernatant, the alginate solution was added to obtain an alginate suspension containing 5 million cells per milliliter. Medium, PBS, Trypsin/EDTA, and the alginate suspension used were warmed to 37°C.
Cell encapsulation by electrohydrodynamic jet method
Cell encapsulation was performed by following a protocol already published. 39 In brief, the encapsulation system was placed under the biological hood, a sterile 3-mL syringe was filled with cells suspended in the alginate solution, connected to the polytetrafluoroethylene tube, and placed onto the pump. The pump was set at the fixed flux of 0.05 mL/min. The other side of the tube was connected to the stainless steel needle. The electrical potential was established by connecting the positive cathode of the generator to the needle and the anode to the metallic plate under the Petri dish. The Petri dish containing the sterile calcium chloride solution was placed below the needle at a distance of 5 cm. Once the flux of solution was established and the generator was switched on (V=8 kV), droplets of alginate solution containing cells were ejected from the needle directly to the CaCl2 solution. After 10 min, the beads were washed thrice with DMEM and redistributed in six-well TCP to perform the experiment.
Cell release
To perform part of in vitro evaluations accordingly to the time points of the experiment, cells were first released from the beads. The hydrogel was dissolved by removing the DMEM containing Ca2+ cations with a short wash in PBS at room temperature, followed by a 15-min incubation in PBS at 37°C to favor the ion exchange with the surrounding buffer. After that, the samples were centrifuged and cells were collected for the experiment.
In vitro evaluation
Cell viability
Cell viability was estimated by fluorescence visualization (confocal microscopy) of samples stained with calcein/propidium iodide. Samples were observed 3 h, 1, 3, 7, 14, 21, and 28 days after encapsulation following standard protocols. In brief, samples were incubated for 20 min at 37°C in a solution of calcein (1 μL of stock solution for 1 mL of DMEM). After that, the supernatant was removed and the beads were washed with a CaCl2 solution and collected from the bottom of the vial after centrifugation (500 rpm for 1 min). The procedure was repeated again at room temperature for 5 min with a solution of propidium iodide (100 μL for 1 mL of CaCl2). To validate the results, a positive control was prepared by inducing cell death with a 5% H2O2 treatment overnight.
Cell proliferation
DNA extraction was performed by placing the cells extracted from the beads in a solution of 0.05% Triton-X in PBS, followed by sonication for 10′′ (cycle: 1, amplitude 40%). After that, DNA was extracted and PicoGreen was used for the quantification. Fluorescence intensity of PicoGreen–DNA complex was measured in 96-well plates on a plate reader (485 nm excitation and 538 nm emission; Safire, Tecan). A calibration curve was built up using the DNA standard provided with the assay to correlate fluorescence intensity to the concentration of DNA.
Gene expression
Gene expression analysis was performed using quantitative reverse transcription polymerase chain reaction (RT-qPCR). Cells were released from the beads, and total mRNA extraction (three replicates for each group) was performed according to the protocol for the QIAGEN mRNA extraction kit. mRNA was resuspended in a final volume of 30 μL with RNAse-free water and its yield was quantified by standard spectrophotometry. Reverse transcription using 1 μg of total RNA was performed with SuperScript III Reverse Transcriptase (Invitrogen) and oligo-dT primers according to the manufacturer's protocol, in a 20 μL volume. Transcribed cDNA was diluted 1 to 20 times till a final volume of 100 μL in RNA-free water was obtained. Primers were selected from the online primer bank PrimerBank. 40 GAPDH primers were designed (Table 1). Reactions were set up in 10 μL volumes, which included 2 μL cDNA, 0.2 μL of each primer, 5 μL of KAPA, and 2.6 μL of RNA-free water. Amplification and detection were carried out using a CFX96 Touch™ Real-time PCR machine (BioRad), using the default universal cycling conditions recommended by the manufacturer. Measurements were performed in triplicate for statistical analysis.
Statistical analyses
All statistical analyses were performed with GraphPad Prism 6 software. Statistical analyses for more than two groups were carried out using one-way analysis of variance with Tukey's post hoc tests. A p value of <0.05 was considered significantly different. Gene expression of SHSY5Y line in TCP (at passage 16) was set up as a control and equal to 1. For gene expression analysis, the ddCT approach was applied. 41 Targeted gene expression was calculated by the fold change related to the control group.
Results
The study was performed up to 4 weeks at different time points: 3 h, 1, 3, 5, 7, 14, 21, and 28 days. During that time, cells self-assembly and viability, proliferation, and gene expression were investigated.
Cell encapsulation
The parameters for the encapsulation process and bead characteristics were already investigated and published. 39 Beads with a diameter of about 200 μm were made using 2% alginate with 8 kV and a 33G stainless steel needle (I.D.: 0.108 mm) (Fig. 1A).

Beads with diameter consistently stable, about 200 μm were made using 2% alginate ejected under 8 kV from a 33G stainless steel needle (I.D.: 0.108 mm) to CaCl2 solution for crosslinking.
Cell self-assembling and migration
Figure 1A (Zeiss optical microscope) represents the morphology, organization, and distribution of cells inside the beads starting from 3 h after encapsulation up to 4 weeks. In the first week, cells were small, round-shaped, and homogeneously distributed at the center of beads without visual changes up to the 7th day. Toward the 14th day, cells started to proliferate and move to the border of the beads. After 2 weeks, cells partially moved outside the beads, adhering to their surface as well as to the TCP. Toward the 28th day after encapsulation, most of the beads were gathered in groups and overpopulated by cells. The beads were distorted and partially degraded in many cases. Cells that migrated and attached to the TCP surface made clusters with the fibroblast-like phenotype from the center to the outside (Fig. 1B).
Cell viability
Calcein/propidium iodide staining followed by fluorescence visualization (Confocal microscopy) was used for the cell viability evaluation. Images showed homogeneous distribution of the cells encapsulated in the beads. The absence of dead cells was observed 3 h after encapsulation, and few 1–3 days later (Fig. 2A). Some dead cells were observed at days 5 and 7. During the following 2 weeks, few beads with only dead cells were observed, but in general there were few dead cells in the beads. In the end, at the 28th day, no dead cells were detected. Results were compared with the positive control samples (necrotic cells).
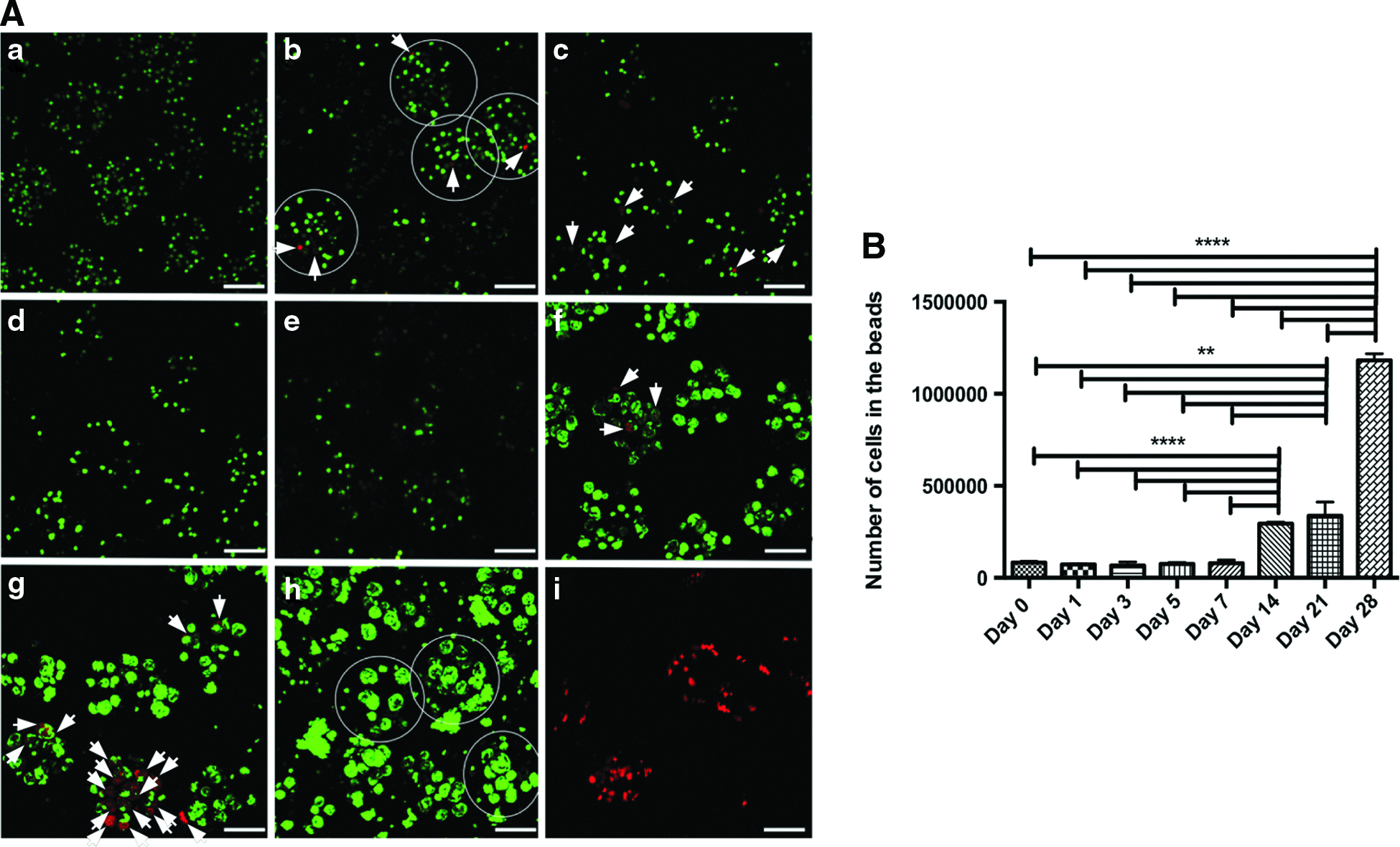
Cell proliferation
Cell viability test results were confirmed followed by DNA quantification assay. Ten percent decrease in cell number was detected 24 h after encapsulation (however, not statistically significant), and no variations in cell number up to the 7th day (Fig. 2B). After the first week, the cell numbers started to increase and tripled from the 3rd to 4th week of encapsulation.
Stress marker expression within the alginate beads
The results of RT-qPCR have been summarized in Figure 3.

Quantitative reverse transcription PCR analysis of stress/apoptotic/necrotic markers in SHSY5Y cells encapsulated in 2% alginate beads by the EHDJ method and evaluated at eight time points. Expression of all targeted genes in studying samples was compared to the negative control samples (cells grown in tissue culture plate) and to the positive control samples (with induced hypoxia, apoptosis, necrosis, and heat shock).34–36
The expression of the targeted genes was compared to cells grown in TCP till 85–90% confluence (culture negative control), and as positive control, cells with induced hypoxia, apoptosis, necrosis, and heat shock were used.34–36 One week after encapsulation, 20 to 80 times increase in HSP70B′ expression was detected, with the highest level after 3 h (no statistical differences from the positive control) (Fig. 3A). However, afterward, a stable decrease in HSP70B′ expression was observed with complete normalization to the negative control level at day 21 after encapsulation. Along with HSP70B′ overexpression, a twofold increase of caspase-3 gene (no difference with the positive control sample) was detected in the first days after encapsulation, followed by stabilization to the negative control level during the next time points, with a slight increase again at day 14. Necrotic marker HMGB1 expression was detected on the negative control level, without statistically different variations during the experimental time points, except at day 14 when it was slightly upregulated and at day 5 when it was downregulated. Expression of both the hypoxic markers HYOU1 and GAPDH was not elevated during the whole encapsulation time. However, HYOU1 expression during the first 3 weeks was significantly below the negative control level, whereas the GAPDH expression was comparable to the negative control samples. In the first 24 h, double upregulation of N-cadherin expression was detected, followed by a significant downregulation at the next time points, stabilization to the control level at days 14 and 21, and downregulation again at day 28.
Discussion
Methods of cell encapsulation are complex, can damage cells, and change their ability to self-assemble or other vital characteristics. A deep and systemic analysis is essential to control cell functionality and activity after processing.
In particular, referring to EHDJ encapsulation, heat, oxygen, or nutrition deficiency, shear stress, as well as high electrical fields can lead to changes in cell behavior, such as decreased proliferation rate and activity, inability of cells to communicate and self-organize, changes in cell molecular mechanisms (cancer metastasis or autophagy), or stress accumulation. Eventually, as a consequence of stress exposure, cell death can be triggered by two different mechanisms: apoptosis or necrosis (Fig. 3). 27 Generally, necrosis is the result of severe trauma or injury, whereas apoptosis or programmed cell death happens as the result of normal cell cycle or can be enhanced by some mild outer stimuli, like hypoxia, heat, or the presence of some molecules. 21 Several methods have been developed for apoptosis/necrosis evaluation, based on the microscopic evaluation of phenotypic changes, nucleus fragmentation, detection of molecular markers, like phosphatidylserine presence, different caspases, and HMGB1 expression or cytochrome c release; some of these methods are mentioned in Figure 4.
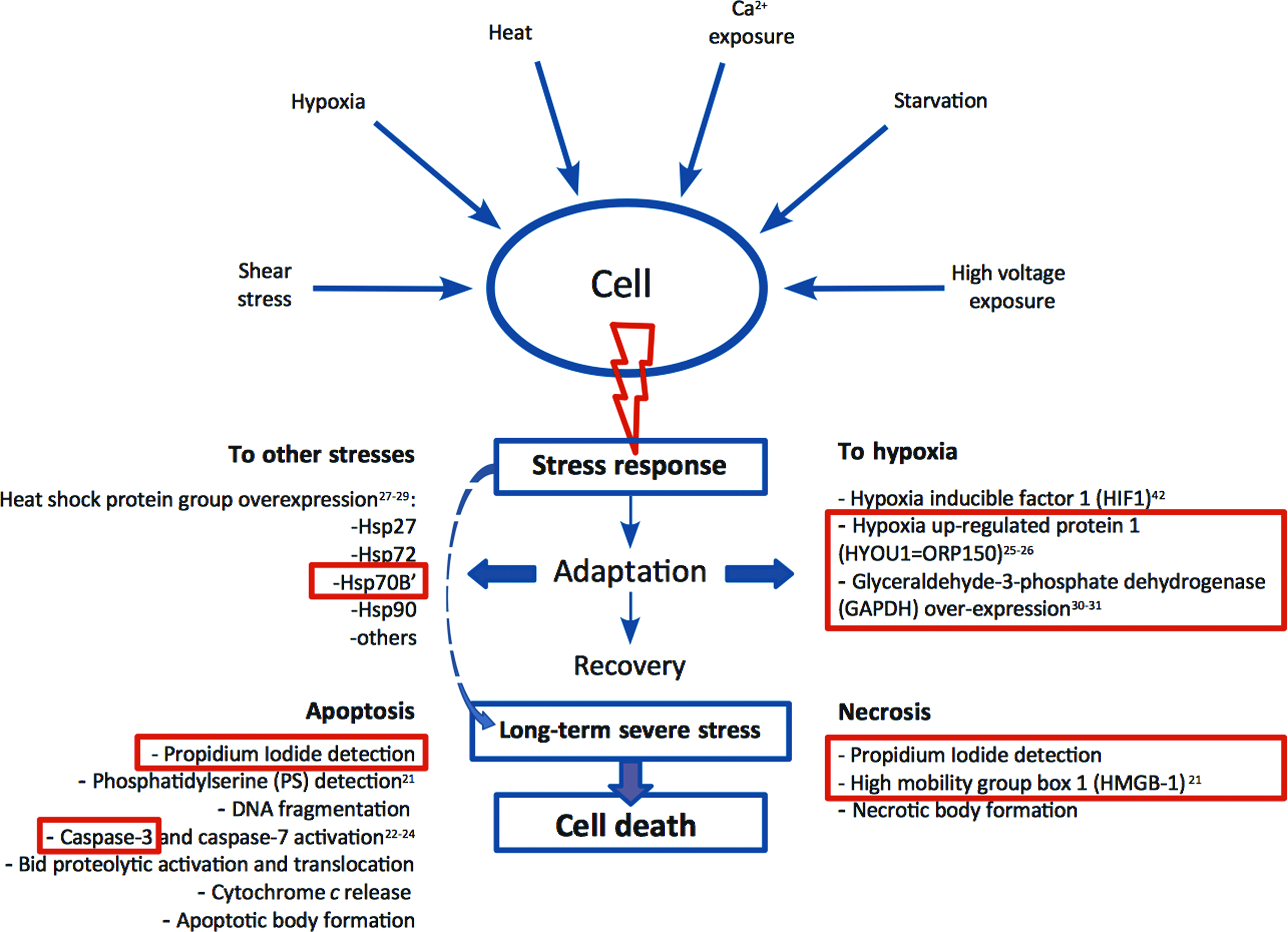
Theoretical model of cell response to stress exposure. In red boxes, factors that were evaluated in this article. Color images available online at
In this study, apoptosis/necrosis detection was performed at the molecular level; methods based on microscopic observations after immunostaining were not efficient and informative due to the interference of the alginate matrix.
Before or in some cases even after the activation of programmed or acute cell death pathways, HSP are produced for protection. 27 The HSP family is composed of proteins that are increasingly expressed in response to stresses (e.g., Hsp70A, Hsp90), with some members of the family specifically expressed in particular stress conditions like heat, hypoxia, and heavy metal exposure (e.g., Hsp60, Hsp70B′, Hsp72).28,29 In literature, there are reports about cell stress resistance due to the HSP overexpression and cell recovery. 27 However, HSP overexpression generally points to the presence of stress with no indications about its nature. In addition to HSP, specific genes like, HIF1α, HYOU1, and GAPDH are good markers for hypoxia detection.25–30,42 Caspase is the major family of proteases that is responsible for apoptosis propagation, with specific genes for each stage: caspase 8,9 are early markers, whereas caspase 3 and 7 are late apoptotic executors, the expression of which can directly point to the propagation of apoptosis.23,24 HMGB1 overexpression is a good diagnostic marker to detect the presence of necrosis.
This study evaluated with a systemic approach the effect of EHDJ encapsulation on the cell characteristics like ability to self-assemble, viability, activity, proliferation, and the expression of specific genes (stress, apoptotic, and necrotic). In vitro evaluations were performed at different time points to check the short-term effect from 3 h up to 7 days after encapsulation and long-term up to 4 weeks. SHSY5Y human neuroblastoma cells at passage 16 were encapsulated in 2% alginate beads (200 μm diameter) applying 8 kV to the extrusion needle.
Initially, the short-term effect (stress molecule production and apoptosis propagation) of EHDJ encapsulation on the cell behavior was analyzed. RT-qPCR analysis indicated that 3 h after encapsulation, the HSP70B′ expression was 80 times higher than in the negative control group (Fig. 3A). The main function of HSP proteins is to protect cells against apoptosis, necrosis, hypoxia, or other type of stresses. Stress accumulation leads to HSP family overexpression, which in turn induces cell stress recovering followed by cell stress resistance.27,38,43 However, notwithstanding the expression of HSPs, under severe stress conditions, cells can overpass the function of HSP and initiate programmed cell death propagation.27–29,38 Cell viability analysis showed the absence of cell death 3 h after encapsulation, however, investigation at the 1st and the 3rd day revealed the presence of some dead cells (Fig. 2A). Cell viability results were confirmed at the molecular level by the 2–2.5-fold increase of the expression of CASP3 (caspase-3) gene in the first hours after encapsulation till the first day (Fig. 3B). In the following days (3–7), CASP3 expression decreased and stabilized to the negative control level, supporting the cell viability test. In fact, at day 7 after encapsulation, a decreased amount of dead cells was observed. Our experimental results agree with the results found in literature about the molecular mechanisms of cell stress protection and apoptosis propagation in stress conditions. 38 Cell death reaches the maximum level 24 h after stress exposure following the caspase-3 cleavage and activation. 44 The DNA quantification test confirmed the above results and showed a 10% decrease in cell number the first day after encapsulation (not significantly different) (Fig. 2B). Up to the 7th day of the experiment, there was not any significant variation in cell number. These results prove that the EHDJ encapsulation system has a mild stress effect on the cells in the first 24 h, however, they are able to recover in a short time and remain viable.
Long-term encapsulation can cause nutrition or oxygen deficiency, which in turn can trigger stress accumulation and apoptosis propagation, as well as hypoxia followed by necrotic cell death. From the 5th day of encapsulation, the level of HSP70B′ increased again up to 50 times. However, after the 7th day, HSP70B′ expression declined in a clear trend with full recovery to the negative control level at day 21. At day 14, together with a slightly elevated HSP70B′ expression, death-related marker levels were also elevated. The cell viability test showed a moderate amount of dead cells (Fig. 2A). Expression of hypoxic markers HYOU1 and GAPDH was comparable to the negative control at later time points (Fig. 3B, C). Up to the 4th week of encapsulation, the expression of stress-related markers was not observed. Along with a decline in the expression of HSP70B′, cells regain their ability to proliferate and the results of DNA quantification test showed a twofold increase in cell number between the first week and the second and triplication toward the 4th week. Presumably, the initial HSP70B′ overexpression led cells to the stress-resistant state, protecting them against hypoxia or other impulses. This is supported by literature and the analysis about stress resistance development after HSP family overexpression in stress conditions.27–29 Peculiarly, a significant decrease was detected in HMGB1 and CASP3 expression at day 5 compared to the negative control. This can be explained by the absence of the physiological 1–5% cell death because of the lack of cell proliferation.21–24 A decreased level of HYOU1 was observed at days 3 and 7. To understand whether HSP70B′ overexpression has a real protective effect on the encapsulated cells, further experiments with HSP70B′ silencing need to be conducted. Obtained results suggest an absence of stress conditions and a stable cell state inside the beads during the 3 weeks followed by the first week of stress recovering after the encapsulation process.
At last, double upregulation of N-cadherin during the first days of the experiment can be the result of cells growing inside the beads. These results suggest that cells released 24 h after encapsulation will have favorable conditions for adhesion and self-assembly, in fact, this phenomenon was described in the literature earlier. 45 However, considering that SHSY5Y cells used in the experiment are tumor cell line, there can be another explanation for this phenomenon. According to the literature review, overexpression of N-cadherin leads to metastatic cell flow and to increased levels of transendothelial migration. 41 However, the chemistry of the material can lead to a decrease in the ability of cells to adhere. This was observed from double downregulation of N-cadherin expression toward the 7th day of encapsulation. During the following weeks, N-cadherin expression was varied, but within the control level. Cells after 2 weeks started massive migration outside the beads. After migration, cells attached to the surface of the alginate beads and the TCP. Alginate lacks adhesion motifs and as a consequence, cell adhesion to its surface was weak. Even though beads were gathered in clusters overgrown by cells, they were not firmly adherent and could be detached applying shaking or pipetting. However, for organ printing application, this technique can be applied in a very efficient way, where cells could be printed in a very precise 3D structure on the top or inside of different matrices, which will support cell migration and self-assembly even better.
Conclusions
In summary, SHSY5Y cells encapsulated by the EHDJ method in 200 μm 2% alginate beads are subject to mild stress conditions, whereby they are able to recover in a short time. In fact, our analysis showed that cells could be encapsulated up to 4 weeks without undergoing cell death or oxygen deficiency. Cells kept their ability to self-assemble in 3D constructs mimicking an in vivo morphogenesis. This is a positive phenomenon for tissue maturation in organ printing application.
This article provides a systemic approach for the evaluation of the effect of EHDJ encapsulation on cell behavior in short and long term (up to 4 weeks after encapsulation). In this study, we proposed a protocol for evaluation of the EHDJ encapsulation effect on the cell behavior. This protocol can be used not only for the EHDJ encapsulation method but also for other tissue engineering methods. To obtain a full picture of the effects of EHDJ encapsulation, further experiments are needed to evaluate tissue/organ maturation in postencapsulation time.6–8
Footnotes
Acknowledgments
The authors acknowledge the Centre of Integrative Biology (CIBIO) of University of Trento for providing the SHSY5Y cell line and Dr. Wei Sun for helping with RT-qPCR measurements and data evaluation.
Disclosure Statement
No competing financial interests exist.