
Abstract
Neural progenitor cells are usually derived from pluripotent stem cells (PSCs) through the formation of embryoid bodies (EBs), the three-dimensional (3D) aggregate-like structure mimicking embryonic development. Cryo-banking of EBs is a critical step for sample storage, process monitoring, and preservation of intermediate cell populations during the lengthy differentiation procedure of PSCs. However, the impact of microenvironment (including 3D cell organization and biochemical factors) of EBs on neural lineage commitment postcryopreservation has not been well understood. In this study, intact EBs (I-E) and dissociated EBs (D-E) were compared for the recovery and neural differentiation after cryopreservation. I-E group showed the enhanced viability and recovery upon thaw compared with D-E group due to the preservation of extracellular matrix, cell–cell contacts, and F-actin organization. Moreover, both I-E and D-E groups showed the increased neuronal differentiation and D-E group also showed the enhanced astrocyte differentiation after thaw, probably due to the modulation of cellular redox state indicated by the expression of reactive oxygen species. In addition, mesenchymal stem cell secretome, known to bear a broad spectrum of protective factors, enhanced EB recovery. Taken together, EB microenvironment plays a critical role in the recovery and neural differentiation postcryopreservation.
Introduction
P
NPCs are usually derived from PSCs through the formation of embryoid bodies (EBs), the aggregate structure mimicking embryonic development.9,14 NPC derivation from PSCs has a lengthy procedure that could last up to 6–14 weeks.10,15,16 Cryo-banking of EBs for NPC derivation provides a necessary step for sample storage, process monitoring, and preservation of the intermediate cell populations. 17 During EB cryopreservation, the 3D cell organization is a critical parameter to maintain the recovered cell properties. 17 For adult neurospheres, disruption of 3D cell organization has been shown to reduce the efficiency of terminal neuronal differentiation.18,19 For PSC-derived NPCs, cryopreservation of the dissociated single cells caused significant apoptosis and required treatment with Rho-associated protein kinase (ROCK) inhibitors or caspase inhibitors to maintain cell viability.11,20 Although cryopreservation of adult neurospheres is feasible, cryopreservation of EBs for neural differentiation has not been well studied. To date, there are only a few studies for cryopreservation of spontaneously differentiated EBs.17,21 Especially, the impacts of EB organization and cryopreservation process on neural lineage commitment of EBs post-thaw have not been fully characterized.
Aggregate-based cryopreservation can preserve cell–cell contact and extracellular matrix (ECM) microenvironment, which are beneficial for cell recovery post-thaw. Cryopreservation of adult NPCs as small intact neurospheres (30–100 μm) resulted in high viability possibly due to the preservation of cell–cell contact. 19 To avoid aggregate fragmentation, encapsulation method was incorporated with slow-cooling procedure to preserve intact neurospheres. 22 Our previous study cryopreserved undifferentiated PSC aggregates in a defined protein-free formulation, 23 which showed that maintaining cell–cell contact and ECM structure could reduce reactive oxygen species (ROS) and caspase expression in small PSC aggregates.23,24 Given the importance of ROS and caspase in regulating cell survival, the secretome of mesenchymal stem cells (MSCs) has also been investigated in our previous study to promote ECM secretion from PSC-derived NPC aggregates. 24 Taking one step further, this study evaluated the cryopreservation effect on the differentiated PSC aggregates (i.e., EBs) for neural lineage commitment.
Specifically, this study investigated the effects of EB structural organization on cell recovery and neural differentiation post-thaw. The hypothesis is that the EB microenvironment and cryopreservation may differentially regulate neural lineage commitment post-thaw due to the modulation of ECMs and cellular redox state. The influence of MSC secretome, known to possess high antioxidant properties, 25 was investigated to modulate oxidative environment of EBs. This study assessed the suitability of cryopreserving EBs and revealed the role of cellular microenvironment on cell recovery and neural lineage commitment after EB cryopreservation and thaw.
Materials and Methods
Undifferentiated ESC culture and generation of EBs
Murine ES-D3 line (Cat# CRL-1934; American Type Culture Collection) was maintained on 0.1% gelatin-coated six-well plates (Millipore) in a standard 5% CO2 incubator. The expansion medium is composed of Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% ESC-screened fetal bovine serum (FBS; Hyclone), 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin (all from Invitrogen), and 1000 U/mL leukemia inhibitory factor (LIF, Cat# ESG1106; Millipore). The cells were seeded at 2–4×104 cells/cm2 and subcultured every 2–3 days.
To generate EBs, ESCs were seeded at 1×106 cells into ultra-low attachment six-well plates (Corning Incorporated) in 3 mL of DMEM-F12 plus 2% B-27 serum-free supplement (Cat# 17504-044; Invitrogen), which was referred as neural differentiation medium. The formed EBs were cultivated for 4 days. At day 4, 1 μM all-trans retinoic acid (Cat# R2625; Sigma-Aldrich) was added in the media to induce neural differentiation. 26 EBs were cultivated for another 4 days and then collected at day 8 (about 300–400 μm in diameter) for cryopreservation study (Fig. 1A).
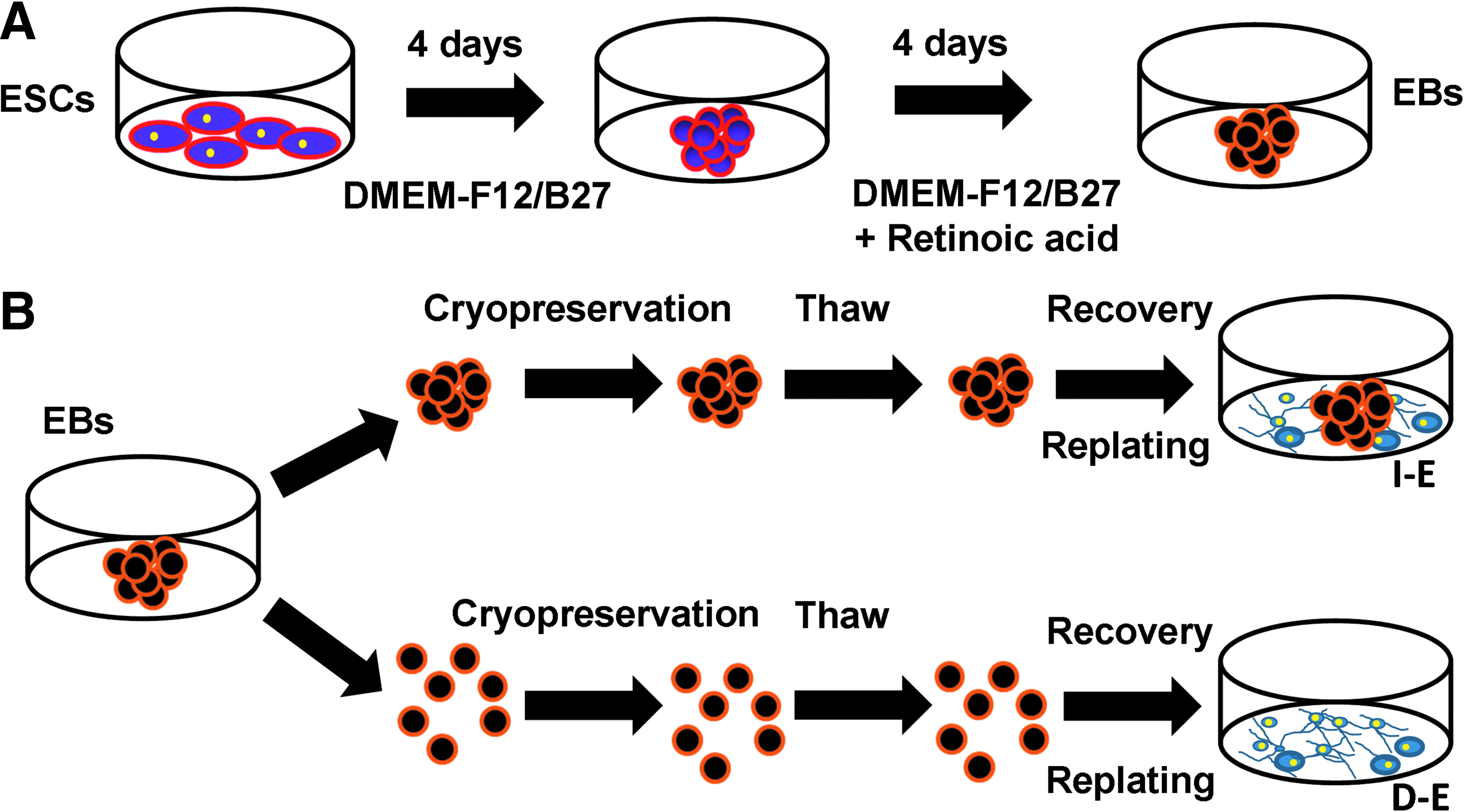
Schematic diagram of the experimental procedure.
Cryopreservation of EBs
The collected EBs were cryopreserved either as intact EB aggregates (I-E) or as single cells after dissociation with 0.05% trypsin/EDTA, that is, dissociated EBs (D-E) (Fig. 1B). For the I-E group, ∼50 aggregates in 250 μL media were taken and dissociated. The resulting single cells were counted by hemacytometer. This number was used to estimate the cell concentration of I-E cells. Based on the calculated cell concentration, different volumes of suspension were taken to obtain the samples with 1×105, 5×105, or 1×106 cells. For the D-E group, a 20 μL of single cell suspension were counted directly. The aggregates or the single cells were washed in phosphate buffered saline (PBS) and resuspended in 90% FBS plus 10% dimethyl sulfoxide (DMSO) (Figs. 2–8) or 90% HypoThermosol® FRS (HTS-FRS; BioLife Solutions) plus 10% DMSO (Supplementary Table S1; Supplementary Data are available online at
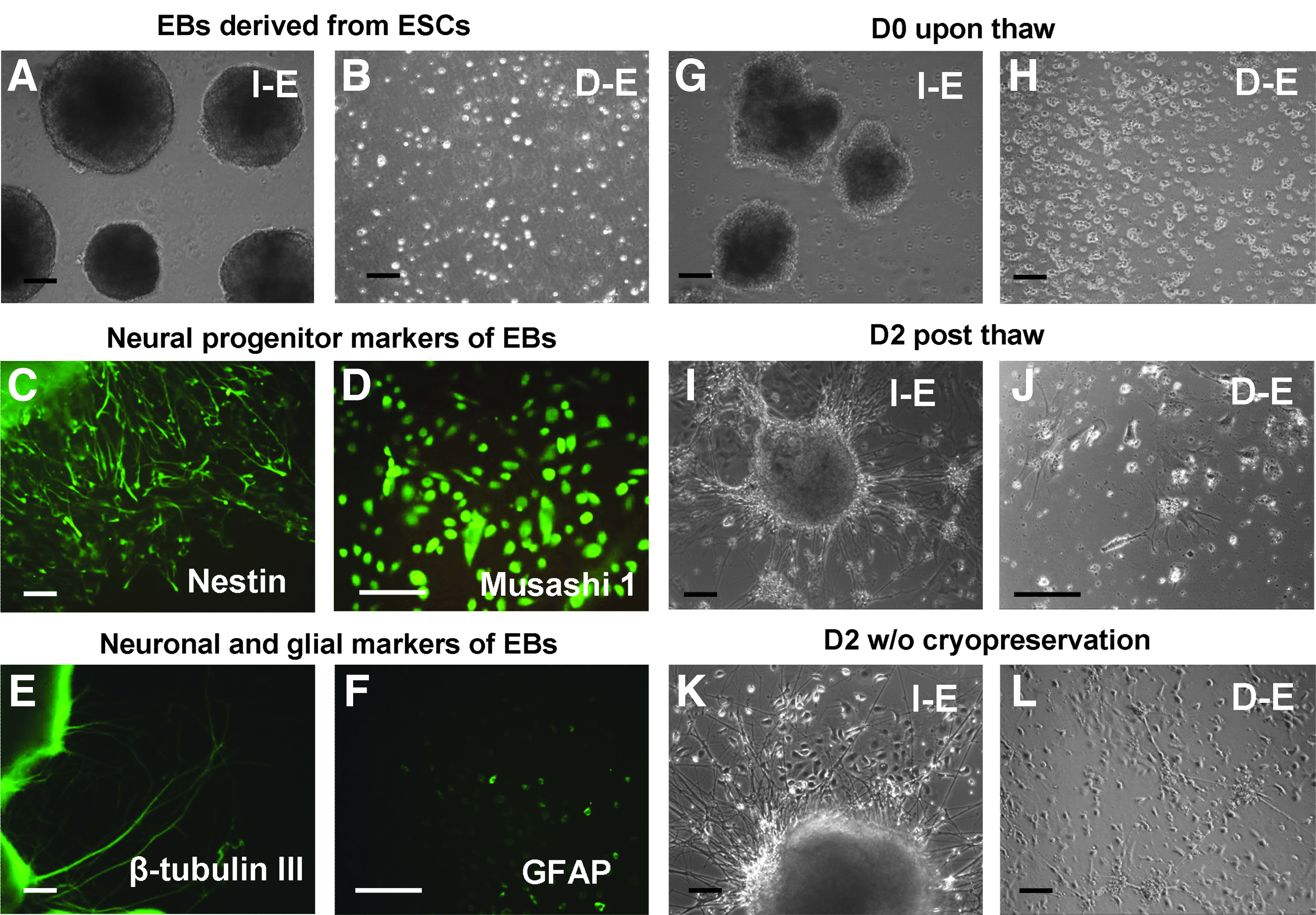
Morphology of EBs before and after cryopreservation. Morphology of EBs before cryopreservation:
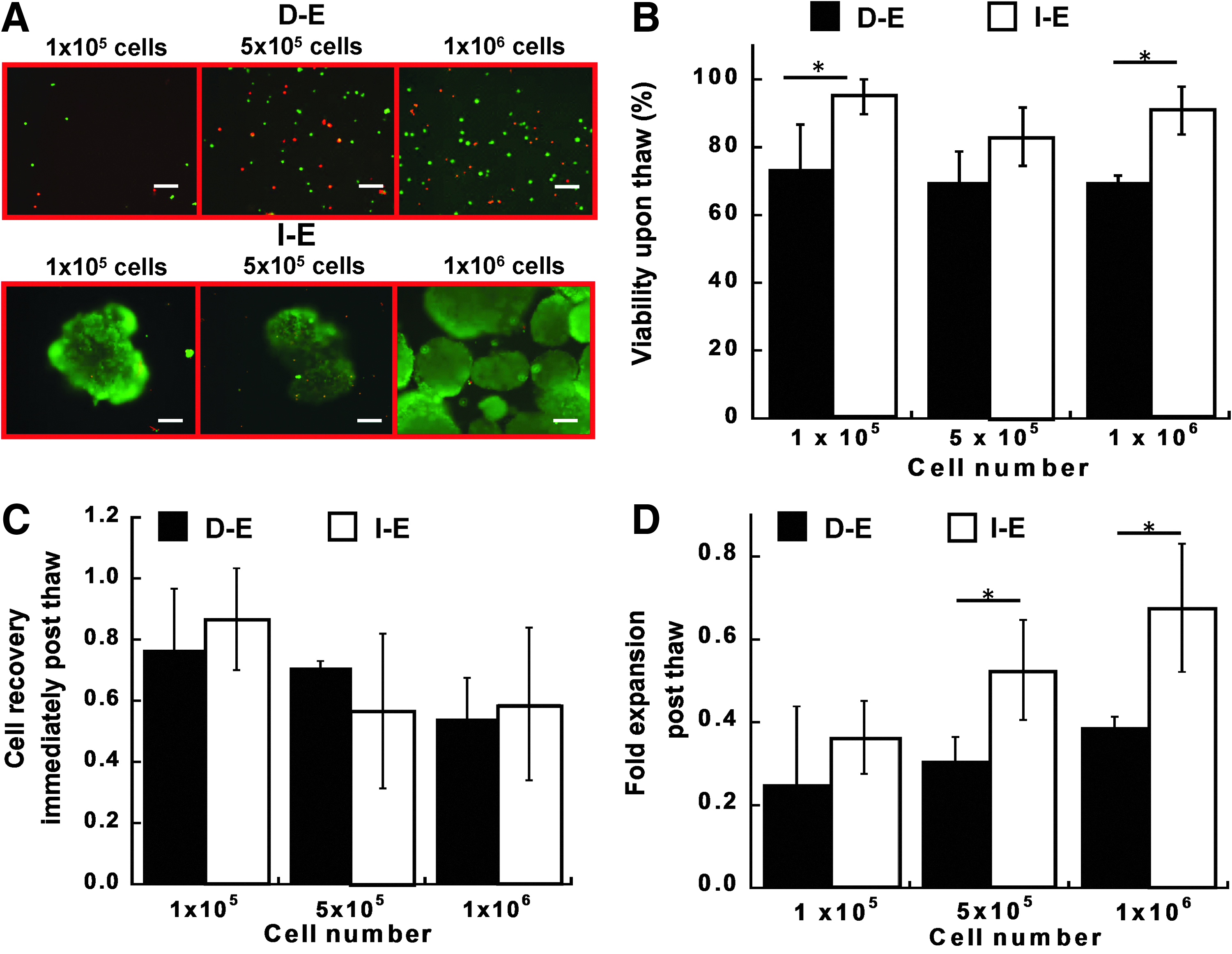
Recovery of intact and dissociated EBs upon thaw and the effects of cell density during cryopreservation.
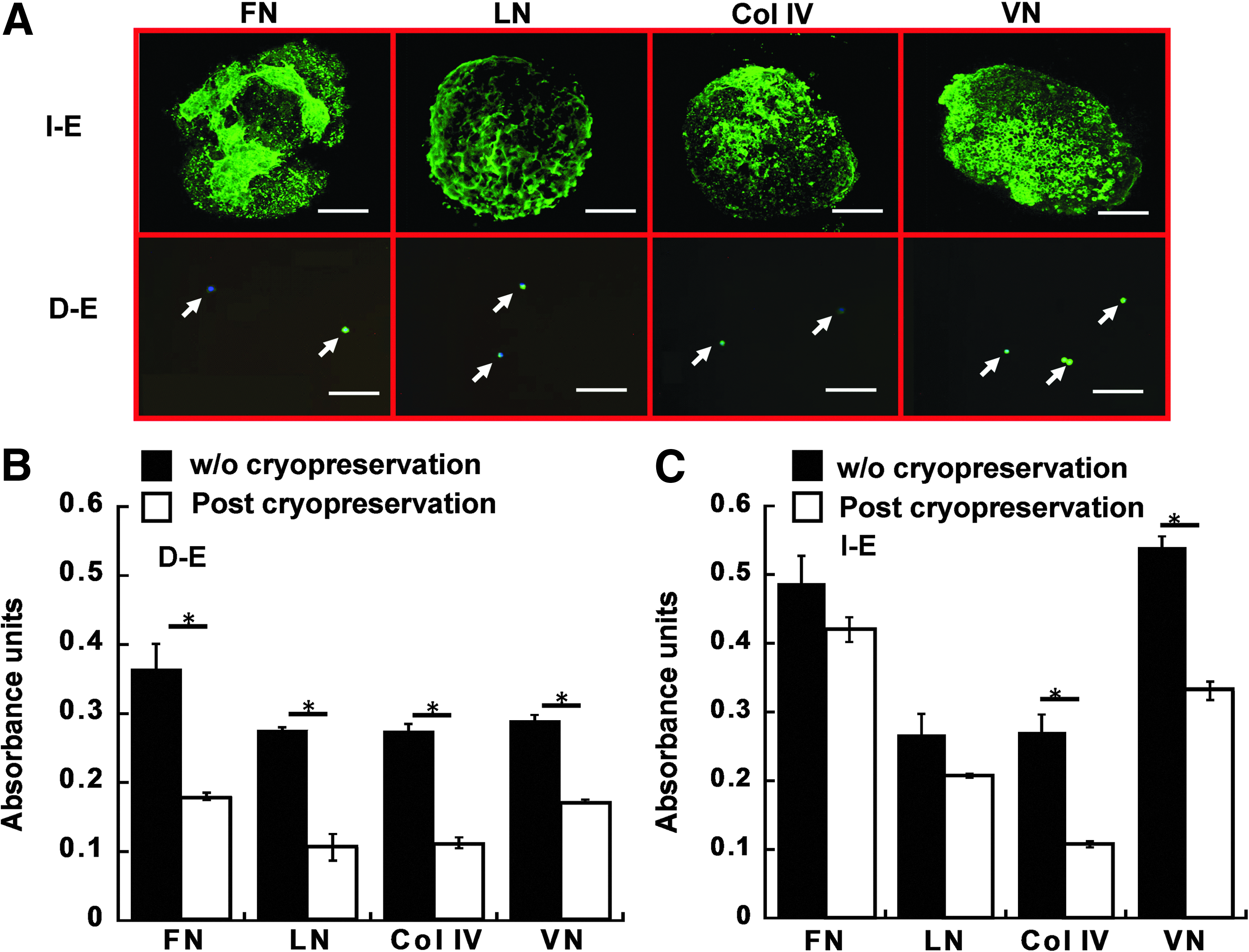
Extracellular matrix expression of intact and dissociated EBs upon thaw.
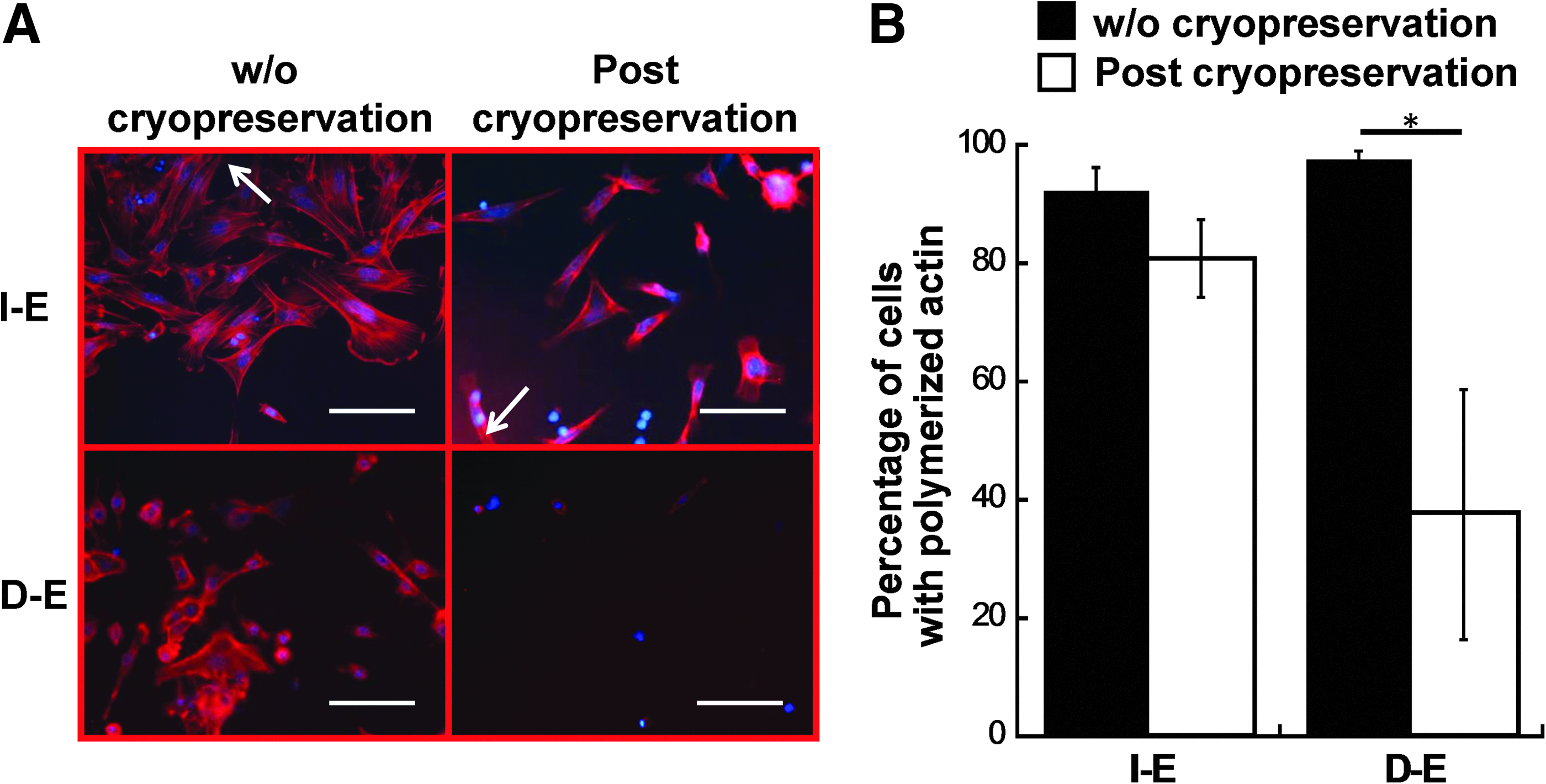
F-actin organization in intact and dissociated EBs after cryopreservation.
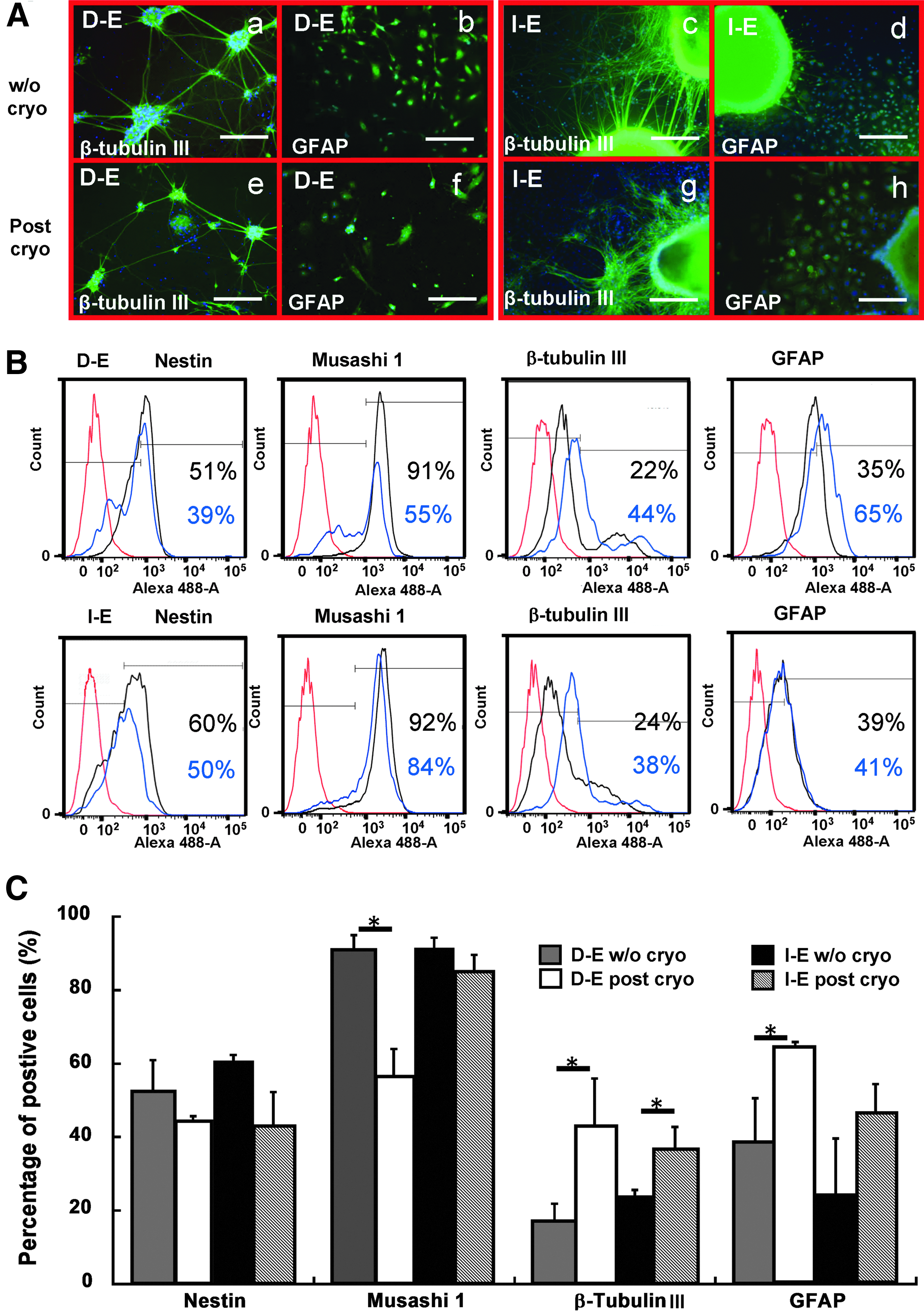
Neural lineage commitment of the intact and dissociated EBs after cryopreservation.
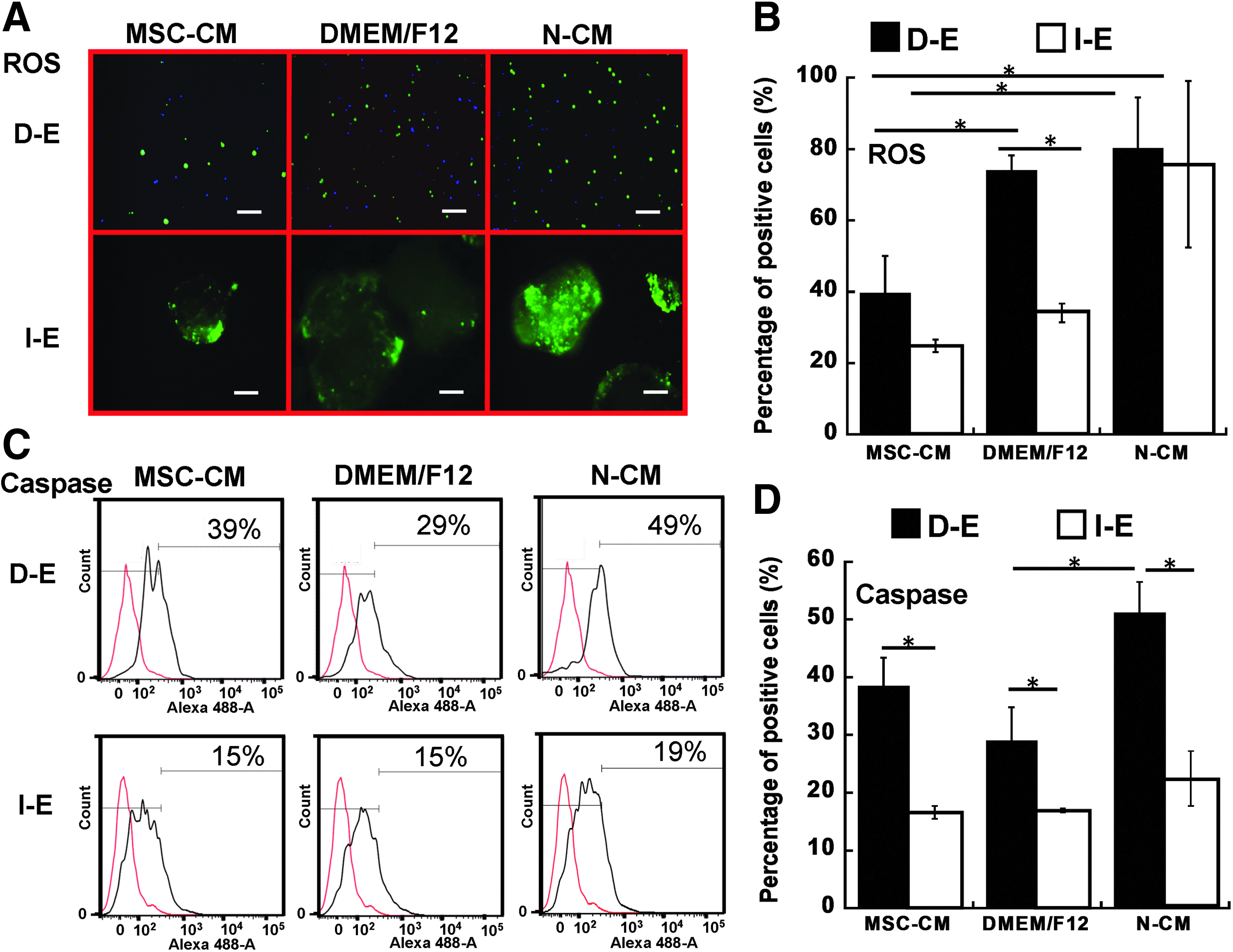
Reactive oxygen species (ROS) generation and caspase activation in EBs after cryopreservation.
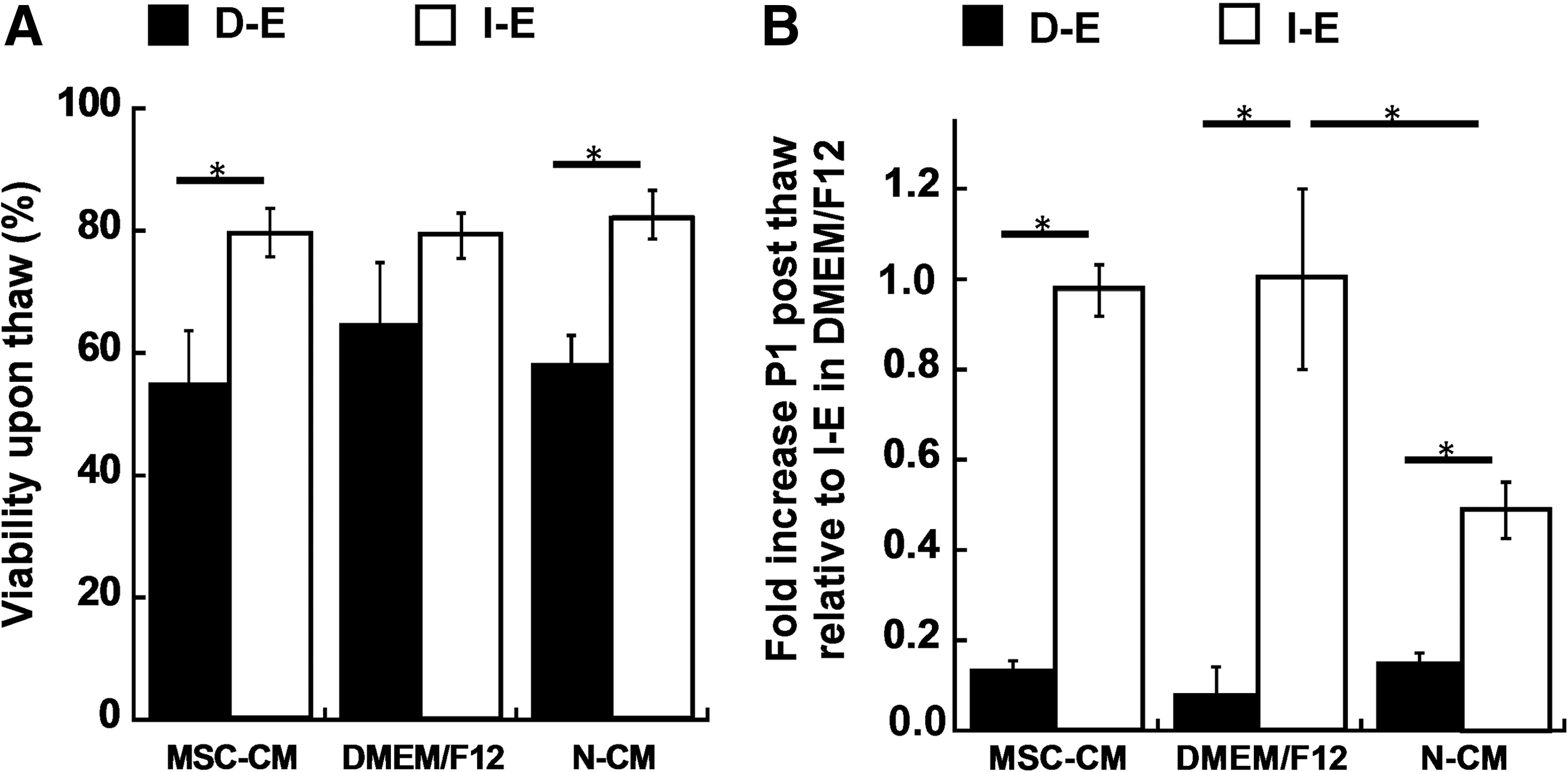
Recovery of intact and dissociated EBs preconditioned in mesenchymal stem cell-conditioned medium (MSC-CM).
Live/Dead, reactive oxygen species, and poly caspase assays
Live/Dead assay
Cell membrane integrity was assessed using LIVE/DEAD® staining kit (Cat# L3224; Molecular Probes). 23 The aggregates and single cells were incubated in DMEM containing 1 μM calcein AM and 1 μM ethidium homodimer I for 30 min. The samples were then washed and imaged under a fluorescent microscope (Olympus IX70) or a confocal microscope (Leica TCS SP2 AOBS). Multiple levels of aggregate analysis showed high viability (Supplementary Fig. S1). Using ImageJ software, the staining intensity (red for dead cells and green for live cells) was measured and each value was subtracted from the background intensity. The viability was calculated as the percentage of green intensity over total intensity.
ROS assay
ROS detection was performed using Image-iT™ Live Green Reactive Oxygen Species Detection kit (Cat# I36007; Molecular probes). 23 Briefly, the I-E and D-E were washed in Hank's Balanced Salt Solution (HBSS) and incubated in a solution of 25 μM carbioxy-H2DCFDA for 30 min at 37°C. The samples were then washed and analyzed under fluorescence microscope. As positive controls, the cells (both I-E and D-E groups) were incubated in a 100 μM tert-butyl hydroperoxide (TBHP) solution, prior to staining with carboxy-H2DCFDA.
Caspase assay
Caspases (caspase-1, -3, -4, -5, -6, -7, -8, and -9) were detected using Image-iT Live Green Poly Caspase Detection Kits (Cat# I35104; Molecular Probes). Caspases-8 and -9 are initiator caspase and caspases-3, -6, and -7 are effector caspase. 27 Briefly, I-E and D-E cells were incubated for 1 h with the fluorescent inhibitor of caspases (FLICA) reagent and analyzed under fluorescence microscope. 23 For quantification, the caspase-stained samples (after dissociation into single cells) were acquired with BD FACSCanto™ II flow cytometer (Becton Dickinson) and analyzed using FlowJo software against the nonstained samples.
ECM expression pre- and postcryopreservation
For ECM expression, cells from I-E and D-E groups were fixed with 4% paraformaldehyde (PFA) immediately upon thaw. The samples were permeabilized with 0.2–0.5% Triton X-100, blocked, and incubated with primary ECM antibodies, including rabbit polyclonal fibronectin (FN, ab23750), laminin (LN, ab11575), collagen IV (Col IV, ab6586), and vitronectin (VN, ab28023) (Abcam). For fluorescence staining, the samples were incubated with Alexa Fluor® 488 goat anti-Rabbit IgG (Molecular Probes), counterstained with 4′,6-diamidino-2- phenylindole (DAPI), and visualized using a fluorescence microscope (Olympus IX70) or a confocal microscope (Leica TCS SP2 AOBS).
To quantify the ECM contents, the same number of cells (1×106) from I-E and D-E groups pre- and postcryopreservation were incubated with donkey anti-Rabbit IgG conjugated with horseradish peroxidase (HRP; Rockland Immunochemicals) after the incubation with primary antibodies. After washing, 1 mL of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Thermo scientific) was added to the samples and incubated for 5–25 min. The reaction was stopped by a solution of 0.16 M sulfuric acid. The absorbance units (AU) were measured using a microplate reader (Biorad) at a wavelength of 405 nm with background subtraction at 655 nm. The AU values were also corrected by subtracting the absorbance of negative control stained with HRP-IgG only. 28
F-actin staining
Actin organization was assessed as reported previously. 29 Cells from I-E and D-E groups before and after cryopreservation were seeded on Geltrex-coated surface for 24 h in neural differentiation medium, fixed with 4% PFA, and permeabilized with 1% Triton X-100 solution. The samples were then incubated for 30 min with Alexa Fluor 594 Phalloidin (A12381; Molecular Probes) and mounted in a mounting solution containing DAPI (Vectashield). The samples were washed and imaged under a fluorescent microscope (Olympus IX70). F-actin polymerization shown as stress fibers and cortical actin was also quantified using ImageJ software.
Immunocytochemistry and flow cytometry analysis of neural markers
After 3 days in neural differentiation medium without any growth factors, the replated D-E and I-E cells were assessed by immunocytochemistry for neural marker expression. Briefly, the cells were fixed in 4% PFA and permeabilized with 0.2–0.5% Triton X-100. The samples were then blocked and incubated with mouse or rabbit primary antibody against Nestin (N5413; Sigma) and Musashi 1 (ab52865; Abcam), both as neural progenitor markers, β-tubulin III (MAB1637; Millipore) as a marker for neurons, and glial fibrillary acidic protein (GFAP, MAB360; Millipore) as a marker for astrocytes. After washing, the cells were incubated with the corresponding secondary antibody: Alexa Fluor 488 goat anti-Mouse IgG1 (for GFAP and β-tubulin III) or Alexa Fluor 488 goat anti-Rabbit IgG (for Nestin and Musashi 1). After washing and mounting in the DAPI-containing solution, the cells were visualized under a fluorescence microscope.
To quantify the levels of differentiation marker expression, flow cytometry analysis was performed. 24 The cells after 5 days were dissociated by trypsin/EDTA into single cells. About 1×106 cells per sample were fixed with 4% PFA and washed with staining buffer (2% FBS in PBS). The cells were permeabilized with 100% cold methanol and blocked using a 5% FBS solution in PBS. The samples were then incubated with primary antibodies against Nestin, β-tubulin III, GFAP, or Musashi 1 followed by the corresponding secondary antibody: Alexa Fluor 488 goat anti-Mouse IgG1 or Alexa Fluor 488 goat anti-Rabbit IgG. The samples were acquired with BD FACSCanto II flow cytometer (Becton Dickinson) and analyzed against isotype controls using FlowJo software. The average values from three independent experiments were presented.
EBs preconditioned with human MSC conditioned medium
Conditioned media from human MSCs (MSC-CM) were kindly provided by the lab of Dr. Teng Ma as previously described. 24 MSC-CM was mixed at 50% v/v with DMEM/F12 plus 2% B27 media to treat EBs, referred as the MSC-CM group. αMEM without conditioning was also mixed with DMEM-F12 plus 2% B27 media for comparison, referred to as the non-conditioned medium (N-CM) group. The DMEM-F12 plus 2% B27 medium was referred as the DMEM/F12 group. I-E and D-E cells were cultivated for 2 days in the presence of three medium conditions before cryopreservation. The viability, ROS and caspase expression of I-E and D-E cells were evaluated upon thaw after cryopreservation. The cells were replated in the medium corresponding to the precryopreservation culture for 3 days and evaluated for fold increase.
Statistical analysis
Each experiment was carried out at least three times. The average values of two or three independent experiments were presented and the results are expressed (mean±mean absolute deviation). In each experiment, triplicate samples were used. To assess the statistical significance, one-way analysis of variance followed by Fisher's LSD post hoc tests were used. A p-value<0.05 was considered statistically significant.
Results
Cellular organization regulated EB recovery
EBs were formed as spherical aggregates and can be dissociated into single cells after the treatment with trypsin/EDTA (Fig. 2A, B). The EBs had abundant expression of neural progenitor markers Musashi 1 (75%±8%) and Nestin (61%±6%), low levels of β-tubulin III (neurons) (11%±4%) and GFAP (astrocytes) (9%±4%) (Fig. 2C–F), and they were capable of differentiating into neural cells. 24 To evaluate the effect of cellular organization, the formed aggregates were cryopreserved either as intact EBs (I-E) or dissociated EBs (D-E). Immediately post-thaw, I-E cells displayed less distinct spherical morphology compared to the noncryopreserved group, while D-E groups remained as single cells (Fig. 2G, H). After 2 days in culture, replated I-E cells displayed dense neurite projections around the aggregates compared to the noncryopreserved group, indicating efficient recovery post-thaw (Fig. 2I, K). Conversely, more debris was observed for D-E group and the replated cells showed more flat and spread morphology compared to the noncryopreserved group (Fig. 2J, L).
The effects of cryopreserved cell density (1×105, 5×105, and 1×106 cells/mL) on cell viability and recovery post-thaw were assessed for I-E and D-E groups. For all the densities, lower cell viability (70–72% vs. 82–96%) was observed for D-E groups compared to I-E groups (p<0.05 for 1×105 and 1×106 cells/mL) (Fig. 3A, B). There was no significant difference in cell viability for the three cell densities within D-E or I-E groups. Immediately post-thaw, the cell number was about 60% of the frozen numbers, indicating the cell loss during the process of loading and unloading cryoprotectant buffer (Fig. 3C). There was no statistical difference in cell recovery between the I-E and D-E groups and among various cell densities. After 3 days in culture, D-E groups showed significantly lower expansion fold compared with I-E groups, especially for high cell density (0.39±0.03 vs. 0.68±0.15 for 1×106 cells/mL) (Fig. 3D). The expansion fold of the recovered cells was slightly lower compared with the noncryopreserved cells (e.g., 0.7–1.1 for I-E cells and 0.4–0.6 for D-E cells). These results indicated that intact EBs improved cell viability and cell recovery post-thaw compared with the dissociated EBs. The high cell density of 1×106 cells/mL was used in the subsequent studies.
Intact EBs preserved ECM expression and F-actin distribution
To assess the preservation of aggregate microenvironment, ECM expressions (including FN, LN, COL IV, and VN) of D-E and I-E cells were determined upon thaw prior to the replating on Geltrex-coated surface (Fig. 4A). From the confocal images, the dense ECM networks of FN, LN, COL IV, and VN were preserved among the cells for I-E group similar to the pattern prior to cryopreservation, while ECM expression in D-E cells was restricted to the cell body. Quantitatively, the expression of all four ECM proteins was significantly decreased by two to three-folds in D-E group after cryopreservation compared with the D-E cells without cryopreservation (Fig. 4B). For I-E group, no significant difference was observed for FN and LN expressions after cryopreservation, but the expressions of COL IV and VN decreased two to three-folds (Fig. 4C). These data indicated that cryopreservation may disrupt ECM structure but intact EBs better preserved ECM expressions.
Because cell–ECM interactions affect cell shape and cytoskeleton organization, the effect of cryopreservation on actin organization of the I-E and D-E cells was also evaluated (Fig. 5). Without cryopreservation, EB-derived cells displayed dominantly cortical actin and a small amount of actin fibers. After cryopreservation and thaw, the cell outgrowth from I-E group displayed similar actin pattern compared to the noncryopreserved cells (Fig. 5A). Consistently, the percentage of I-E cells with polymerized actin was similar (92%±4% vs. 98%±2%) for the cryopreserved and noncryopreserved cells (Fig. 5B). In contrast, after cryopreservation, D-E cells showed disorganized actin and the percentage of cells with polymerized actin dramatically decreased from 81%±7% to 37%±21% compared with the noncryopreserved group. These results indicated that intact EBs better preserved actin organization.
Cell organization and cryopreservation regulated EB neural differentiation
The effects of cell organization and cryopreservation on neural differentiation of the replated EBs were evaluated. For the cells in D-E group, the thawed cells differentiated into β-tubulin III+ neurons with extensive neural network (Fig. 6A). More GFAP-positive cells in the cryopreserved group were also observed compared to the corresponding noncryopreserved group. Consistently, flow cytometry analysis indicated that the percentages of β-tubulin III+ cells (17%±5% vs. 43%±13%) and GFAP+ cells (39%±12% vs. 64%±2%) for the cryopreserved cells were significantly increased compared with the noncryopreserved cells (Fig. 6B, C). In the meanwhile, the expression of Musashi 1 was significantly decreased after cryopreservation (91%±4% vs. 56%±8%), while Nestin expression decreased slightly (53%±8% vs. 45%±1%).
For the I-E group, the cells displayed a dense amount of neurons positive for β-tubulin III around the aggregates after cryopreservation (Fig. 6A). Slight increase in GFAP+ cells was observed. The quantitative analysis indicated the significant increase of β-tubulin III+ cells (24%±1% vs. 37%±6%) after cryopreservation (Fig. 6B, C). The increase in GFAP+ cells (25%±15% vs. 46%±8%) was also observed, but the difference was not statistically significant. Musashi 1 expression was comparable with or without cryopreservation (91%±3% vs. 85%±5%), while Nestin expression decreased slightly after cryopreservation (61%±2% vs. 43%±9%). These data indicate that cryopreservation promoted neuronal differentiation of both intact and dissociated EBs, while the EB dissociation in combination with cryopreservation promoted glial differentiation.
Regulation of ROS expression and caspase activation
Because cryopreservation induces oxidative stress, the ROS and caspase expressions were evaluated for the cells in D-E and I-E groups. The cells preconditioned with MSC-CM was also evaluated due to the antioxidant effects of MSC-CM. 24 MSC-CM and N-CM contained 1% B27 due to the 1:1 mixing of EB medium and MSC medium, compared to DMEM/F12 with 2% B27 (control). For the DMEM/F12 condition, the cells in I-E group showed lower ROS expression (34%±3% vs. 74%±4%) compared with the D-E cells (Fig. 7A, B). Preconditioning with MSC-CM reduced ROS expression from 74%±4% to 39%±10% for D-E cells and from 34%±3% to 24%±2% for I-E cells, indicating better antioxidant capability of I-E cells than D-E cells. To interrogate potential contribution of ROS expression to apoptosis, the level of caspase expression was determined (Fig. 7C, D). I-E cells showed significantly lower levels of caspase in all three medium conditions than D-E cells (17–23% vs. 29–51%). Preconditioning with MSC-CM reduced caspase expression compared with N-CM group, but the levels were comparable to the expression in DMEM/F12 group.
To investigate the potential benefit of using MSC-CM preconditioning to improve cell recovery, cell viability and expansion fold were evaluated after cryopreservation. Cell viability immediately upon thaw was similar in all three medium conditions for D-E or I-E cells (Fig. 8A). But the expansion fold was significantly increased (two-fold) in MSC-CM compared with N-CM for I-E cells, and it was comparable to that in DMEM/F12 group (Fig. 8B). While MSC-CM had protection effects, the impact of EB organization (intact vs. dissociated) on cell expansion fold was more pronounced as seen in Figure 8.
Discussion
EB cryo-banking enhances the capacity of basic research and cell therapy by providing a large quantity of cells at the intermediate stage over the course of differentiation, which can be used for process monitoring, sample storage, and follow-up differentiations.13,17 Various cryopreservation methods including slow-cooling and vitrification have been tested for aggregate-based stem cells.19,20,30,31 Slow-cooling method is more amendable for large-scale cell banking due to its simplicity.32–34 However, the impacts of microenvironment on the lineage commitment of cryopreserved EBs using slow-cooling method are not fully understood. This study revealed the influences of endogenous ECM secretion, actin cytoskeleton organization, and cellular redox state (indicated by ROS generation) in EBs on cell recovery and neural differentiation post-thaw.
The microenvironment of EBs regulated cell recovery
The preservation of intact EB microenvironment enhanced cell recovery due to the preservation of cell–cell contacts, endogenous ECM network, and organized distribution of actin cytoskeleton.35–37 The disrupted cell–cell contacts and the cell detachment from the surrounding ECM was reported to result in a specific type of apoptosis, known as “anoikis.” 38 NPCs upon dissociation were prone to apoptosis through the activation of ROCKi, 39 while abundant N-cadherin maintained in cell–cell contacts of intact aggregates was reported to enhance NPC attachment by acting as anchoring points for actin organization.40,41 The process of cryopreservation can result in actin disorganization and the disruption of cadherin and connexin,42–45 impacting stem cell proliferation and survival. However, simply adding ROCK inhibitor Y27632 in the dissociated EBs did not significantly improve the recovery (Supplementary Fig. S2). Instead, intact EBs preserved ECM expression and maintained proper F-actin organization due to the preservation of cellular organization.
Moreover, cryopreservation generates ROS due to the oxidative stress caused by hyperthermia.23,46 High concentration of ROS activated caspase expression and induced cell apoptosis during cryopreservation. 27 In this study, intact EBs had lower ROS expression and less caspase activation compared to dissociated EBs, which may be attributed to the preserved ECM expression. The endogenous ECMs have been reported to mediate antioxidant effects and reduce the intracellular levels of ROS, improving stem cell survival and proliferation.47,48 The sugar components of the ECMs (e.g., glycosaminoglycans) also can exert cryo-protective effects (e.g., mimicking the protection effect of trehalose). 17 In this study, intact EBs retained expressions of laminin and fibronectin, which may contribute to better cell survival. 49 Therefore, the microenvironment of intact EBs may protect the cells from oxidative stress.
Diffusion limitation has been an issue for aggregate-based cryopreservation.23,50 The size of neurospheres and the PSC aggregates reported in previous studies were about 100–200 μm in diameter to allow the efficient diffusion of cryoprotectants and antioxidants. Our previous study has shown that ROS generation and caspase activation were elevated for large undifferentiated PSC aggregates (>300 μm).19,23 In this study, the size of EBs after 8-day culture was around 400 μm, which was inductive for ROS generation. The survival of large EBs could be due to the intrinsically high antioxidant capacities of neural lineages (e.g., expression of antioxidant enzymes: peroxide dismutase and catalase). 51 The cellular redox state, a balance between the rates of production and the clearance of the free radicals, was shown to be better controlled in neural progenitors, which rendered the cells more resistant to oxidative stress. 51
Secretome of MSCs has been shown to exert antiapoptotic and antioxidative effects on neural cells due to factors such as stanniocalcin-1 (STC-1), 25 vascular endothelial growth factor (VEGF), and stromal cell-derived factor-1 (SDF-1).52,53 In this study, MSC-CM was shown to reduce the ROS expression and can be potentially used to improve EB recovery compared to the nonconditioned media. However, MSC-CM did not impact the cell viability immediately post-thaw but the recovery after 3 days. It was postulated that MSC-CM may protect the cells from the delayed-onset cell death and stimulate cell proliferation post-thaw.24,54
Cryopreservation and cell organization modulated neural differentiation
In this study, an increased neuronal differentiation (13–26%) indicated by β-tubulin III expression was observed upon thaw for both intact and dissociated EBs compared with noncryopreserved groups. Although the cryopreservation of spontaneously differentiated EBs has been evaluated, no quantification has been performed for lineage-specific differentiation.17,21 For undifferentiated human ESCs, cryopreservation has been reported to diminish Oct-4 expression and induce spontaneous differentiation, 55 while the impact of cryopreservation on neural differentiation of EBs remains unknown. The increased neuronal differentiation observed in this study is thought to be due to cryopreservation-induced ROS generation. Besides the impact on cell survival, ROS is also reported to serve as a regulator of neural lineage commitment. 56 For example, endogenous ROS was shown to regulate the proliferation and the expression of progenitor markers (e.g., Nestin and doublecortin) for adult NPCs.57,58 The induction of ROS at the stage of proliferation or formation of neurospheres was found to promote neuronal differentiation by activating the phosphoinositide 3-kinase (PI3K)/AKT, p38 mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinases (ERK) signaling pathways.56,57 Our results indicated that cryopreservation may slightly bias the lineage-specific differentiation of PSCs, which needs to be considered for the design of EB cryo-banking process.
Glial differentiation was also promoted after cryopreservation, especially for the dissociated EBs (by 25%), which may be attributed to both ROS generation and cell organization. When ROS were elevated during NPC differentiation, it has been reported that cell proliferation was decreased and glial differentiation (i.e., GFAP+ cells) was promoted at the expense of neuronal differentiation (i.e., β-tubulin III+ cells).59,60 From our results, the glial lineage differentiation was more pronounced in dissociated EBs than intact EBs after cryopreservation, which may be due to the differences in cellular organization of single cells versus multicellular aggregates. For example, the connexin disruption of neural progenitors has been shown to reduce neuronal differentiation but increase glial differentiation. 61 Hence, cellular organization of EBs not only affected EB recovery, but also impacted differentiation potential postcryopreservation.
Cellular redox state reflected by ROS generation is capable of balancing between self-renewal and differentiation of progenitor cells.57,59 Several factors in this study contributed to the regulation of cellular redox state in EBs, including endogenous ECMs, cell organization/cytoskeleton, and the cryopreservation/thaw process. The interplay of these multiple factors impacted EB recovery and the neural lineage specification. While the results of this study indicated the importance of EB microenvironment in regulating cellular redox state, the exact mechanism of cryopreservation-induced neural differentiation of EBs and the impacts on mesoderm and endoderm differentiation still need to be further investigated.
Conclusion
The preservation of cellular microenvironment of EBs protected the cells from cryo-injuries and enhanced cell recovery compared with the dissociated cells, which may be due to the maintenance of proper cell–cell contacts and endogenous ECMs. In addition, the structural organization of EBs and the elevated ROS due to cryopreservation modulated neural and glial differentiation of EBs, which resulted in the increased neuron marker expression and the differential effect on astrocyte marker expression. The results indicated that modulating EB microenvironment prior to cryopreservation affected the differentiation potential, which needs to be considered during cryo-banking of EBs for their applications in drug discovery and regenerative medicine.
Footnotes
Acknowledgments
The authors would like to thank Ms. Ruth Didier of FSU Department of Biomedical Sciences for her help in flow cytometry and confocal microscopy and Ms. Yijun Liu of Department of Chemical and Biomedical Engineering for providing conditioned medium of mesenchymal stem cells. This work is supported by FSU startup fund, FSU GAP award, and partially from the National Science Foundation (grant No.1342192). We also thank Dr. Teng Ma of Department of Chemical and Biomedical Engineering at FAMU-FSU College of Engineering for comments on the article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.