
Abstract
Intestinal in vitro models are valuable tools in drug discovery and infection research. Despite several advantages, the standard cell line-based Transwell® models based for example on colonic epithelial Caco-2 cells, lack the cellular complexity and transport activity associated with native small intestinal tissue. An additional experimental set-back arises from the most commonly used synthetic membranes, on which the cells are routinely cultured. These can lead to an additional barrier activity during in vitro testing. To overcome these limitations, we developed an alternative primary human small intestinal tissue model. This novel approach combines previously established gut organoid technology with a natural extracellular matrix (ECM) based on porcine small intestinal scaffold (SIS). Intestinal crypts from healthy human small intestine were expanded as gut organoids and seeded as single cells on SIS in a standardized Transwell-like setting. After only 7 days on the ECM scaffold, the primary cells formed an epithelial barrier while a subpopulation differentiated into intestinal specific cell types such as mucus-producing goblet cells or hormone-secreting enteroendocrine cells. Furthermore, we tested the influence of subepithelial fibroblasts and dynamic culture conditions on epithelial barrier function. The barrier integrity was stabilized by coculture in the presence of gut-derived fibroblasts. Compared to static or dynamic culture on an orbital shaker, dynamic culture in a defined perfusion bioreactor had an additional significant impact on epithelial cell differentiation, indicated by high prismatic cell morphology and upregulation of CYP3A4 enzyme and Mdr1 transporter activity. In summary, more physiological tissue models as presented in our study might be useful tools in preclinical research and development.
Introduction
I
Mimicking the natural microenvironment in the gut leads to more physiological and predictive in vitro models, which may help to reduce animal experimentation and to counteract the rising failure rates in mandatory phase of clinical trials. The most widely used in vitro test systems rely on immortalized human adenocarcinoma cell lines such as Caco-2 or T84.3–6 These cells are commonly grown on semi-permeable Transwell® inserts, where they form a polarized monolayer and exhibit microvilli formation. Despite many advantages such as easy cell culture and high reproducibility, these models have severe limitations in regards to properties required for a successful drug absorption test system. These limitations include for example, the lack of important mucus-secreting goblet cells and inconsistent expression of drug transporters and metabolic proteins depending on the culture conditions applied. In addition, tight junction formation between Caco-2 cells lead to an unusual tight barrier as exemplified by transepithelial electrical resistance (TEER) values greater than 300 Ωcm2. 7 These TEER values are rather artificial high when compared to the native human small intestine with ∼40 Ωcm2. 8
Recent developments in terms of cultivating human primary intestinal epithelial cells (hIECs) enabled the development of novel, more in vivo-like organoid models.9,10 These organoids are small representatives in structure and function of the respective native organ. Adult intestinal (stem) cells directly isolated from human biopsies can be reliably propagated within this system for several months without significant changes in phenotype. Subsequently, these cells can then be reliably differentiated into the main intestinal cell types more closely mimicking the tissue development in vivo. 9 However, the reduction of high costs and the complex handling of this kind of organoid culture still remain challenging. Moreover, such systems cannot be used to assess classical functionality that is typically determined in two-dimensional Transwell cultures.
Basic requirements, such as the formation of monolayers with tight junctions, intestinal permeability, and transport cannot be provided by the organoid culture. 1 To overcome some of these pitfalls, our study combines intestinal human primary organoids with a Transwell-like approach. Here, we cultivated organoid-derived cells on a decellularized natural matrix prepared from porcine gut (small intestinal submucosa [SIS]). The scaffold is composed of conserved extracellular structures and basal lamina components. 11 Such matrix resembles a much closer physiological 3D microenvironment and entails no additional barrier, which is of relevance especially for high-molecular test compounds or nanoparticles. Since shear stress and coculture with feeder cells have been shown to promote epithelial barrier function,11,12 we also integrated such advanced culture conditions in our primary Transwell-like tissue model. Taken together our model might provide a promising and reliable in vitro test system of the small intestine (Fig. 1).

Schematic of primary intestinal tissue model established in this study. IECs were isolated from human small intestinal tissue and expanded as organoid culture for 3–4 weeks. After enzymatic digestion single cells were seeded on a decellularized biological scaffold (SIS) with or without intestinal fibroblasts. Tissue models were cultured for 7 days under static or dynamic conditions and can be used for various research applications (Illustration gut:
Materials and Methods
Human tissue
Full-thickness jejunal tissues for hIECs and fibroblasts isolation were obtained from obese patients undergoing a stomach bypass operation at the surgery unit, of the University Hospital Würzburg (n = 6, mean age 43.5 years, two male/four female). Informed written consent was obtained beforehand and the study was approved by the Institutional Ethics Committee on human research of the Julius-Maximilians-University Würzburg (study approval number 182/10). The data were analyzed anonymously and according to the principles expressed in the “Declaration of Helsinki.”
Isolation and culture of human IECs and fibroblasts
IECs and fibroblasts were isolated from human full-wall gut resections, 10 cm2 in size as described previously.9,13 Briefly, villi were scraped off the muscle-free mucosa using a sterile glass slide. The remaining tissue was transferred into a 50 mL falcon tube with 20 mL 4°C cold HBSS− (Sigma-Aldrich, St. Louis, MO), vortexed for 5 s and the supernatant discarded. This washing step was repeated until the supernatant was completely cleared of cell debris. Afterward, the tissue was incubated in 4°C cold 2 mM EDTA/HBSS− solution (Sigma-Aldrich, St. Louis, MO) for 30 min at 4°C under gentle rotation on a shaker. Subsequently, the tissue was washed once in 20 mL HBSS− by manually inverting the tube five times. The mucosa was transferred in a new tube with 10 mL HBSS− and manually shaken five times. This shaking procedure was repeated four times always using a new tube. Each cell fraction was checked for the amount and size of crypts within small drops under the microscope. The supernatants containing the most vital appearing crypts were pooled and centrifuged at 350 g for 3 min at room temperature (RT). Pellet was resuspended in 10 mL basal medium, DMEM-F12 Advanced (Invitrogen, Carlsbad, CA) supplemented with 1× N2, 1× B27, 1× Anti-Anti, 10 mM HEPES, 2 mM GlutaMAX-I (all from Invitrogen, Carlsbad, CA), 1 mM N-acetylcysteine (Sigma-Aldrich, St. Louis, MO), and the crypt number was estimated in a 10 μL drop by microscopy. Crypts were centrifuged in a nonstick 1.5 mL tube at 350 g for 3 min at RT and the supernatant was removed. The tube with the cell pellet was placed on ice until further use. The pellet was resuspended in an appropriate amount of cold Matrigel® (Corning, Hickory, NC) that is, ∼5000 crypts/mL. Drops of 50 μL per well were seeded in a 24-well plate and incubated for 10–20 min until the Matrigel was well solidified. The culture medium contained a mixture of 50% fresh basal medium and 50% Wnt3A-conditioned medium. 14 Furthermore, the following growth factors were added: 500 ng/mL hR-Spondin 1 (PeproTech, Rocky Hill, NY), 100 ng/mL mNoggin (PeproTech, Rocky Hill, NY), 50 ng/mL mEGF (PeproTech, Rocky Hill, NY), 10 μM Y-27632 (ROCK inhibitor; Tocris Bioscience, R&D Systems, Minneapolis, MN), 10 nM Gastrin ([Leu15]-Gastrin I; Sigma-Aldrich, St. Louis, MO), 10 mM Nicotinamide (Sigma-Aldrich, St. Louis, MO), 500 nM A83-01 (Tocris Bioscience; R&D Systems, Minneapolis, MN), 10 μM SB202190 (Sigma-Aldrich, St. Louis, MO), 500 nM LY2157299 (Axon MedChem, Groningen, The Netherlands), and 500 μL of this mixture was added per well. Organoids were harvested by resuspension of the Matrigel in 500 μL of Cell recovery solution (Corning, Hickory, NC) per well and incubated for 1 h on ice. For single cell expansion, organoids were digested with TrypLE Express (Invitrogen, Carlsbad, CA) at RT for 10 min and seeded again in Matrigel with readjusted cell number of about 500 cells per Matrigel drop. Additional, 10 μM JAG-1 (Anaspec, Fremont, CA) and 10 μM Y-27632 were added for the first 2 days. To enrich the stem cell population organoids were passaged every 5–7 days. 1
Subepithelial fibroblasts were isolated after IECs processing by digestion of the remaining connective tissue. Connective tissue was cut into small pieces with sterile scissors and transferred to a tube with 0.3 mg/mL Dispase II (Invitrogen, Carlsbad, CA)/0.25 mg/mL Collagenase Type XI (Sigma-Aldrich, St. Louis, MO) solution. After digestion for 30 min at 37°C the cells were centrifuged and the pellet resuspended in medium. Cells were cultured in FibroLife (CellSystems, Troisdorf, Germany) supplemented with 2% fetal bovine serum (Bio&Cell) and Anti-Anti (1 × ). Fibroblasts were used for Transwell-like models up to passage 5.
In vitro Transwell-like models
Primary models of the intestinal mucosa were prepared by seeding 8 × 105/cm2 single IECs with or without 4 × 105/cm2 fibroblasts on decellularized porcine gut scaffolds (SIS) fixed between two plastic cylinders, so-called cell crowns and cultured in 24-well plates at 37°C in the incubator. Seeded area of the 24-well format is 0.54 cm2 per model. For SIS preparation, porcine jejunal segments were explanted from 6-week-old domestic pigs (Niedermayer, Dettelbach, Germany) and decellularized according to a standardized protocol published previously.15,16 Models were cultured for 7 days either under static conditions in a standard well plate, under dynamic conditions on an orbital shaker (OS, 77 rpm) or in a perfusion bioreactor (BR, flow rate: 3.8 mL/min). All models were initially kept in proliferation medium for 48 h under static conditions allowing cells to adhere. Afterward, medium was changed to differentiation medium, that is, reduction of Wnt3a-conditioned medium to 25% and no addition of nicotinamide and SB202190. Additionally, 10 μM DAPT (N-[N-(3,5-Difluorophenacetyl)-
Design of culture systems and fluid dynamic simulation
The cell crown system and the bioreactor were designed to allow the culture of tissues at the interface between two separated fluid domains. For the design of tailored parts, SolidWorks 2014 x64 Edition (Dassault Systemes Deutschland GmbH, Stuttgart, Germany) was employed. Construction drawings were forwarded to GT Labortechnik (Arnstein, Germany) and parts were manufactured from polyether ether ketone (PEEK). To characterize the fluid dynamics, both culture systems were investigated by finite element method computations. Therefore, COMSOL Multiphysics (Comsol Multiphysics GmbH, Berlin, Germany) was employed. Geometry data were imported into the simulation software and the fluid domains were parameterized as water at a temperature of 37°C. Furthermore, the fluid density was set to 1005.5 kg/m3 and the kinematic viscosity was set to 7.65 × 10−3 Pa × s.
Histologic and immunofluorescence analyses
Tissue and scaffold samples were fixed with 4% PFA for 1 h at 4°C, processed for embedding in paraffin, and sectioned with a thickness of 5 μm on a microtome (SM2010 R; Leica, Germany). Tissue slices were first deparaffinized with xylene and rehydrated in a graded series of ethanol according to standard protocols. Tissue and scaffold sections were stained with hematoxylin and eosin (H&E, Morphisto, Frankfurt am Main, Germany), Feulgen (Merck, Darmstadt, Germany), and Masson-Goldner trichrome (Chroma, Muenster, Germany) according to standard protocols for general morphological investigation.20–22
Further characterization of the models was done by immunofluorescence staining. Briefly, antigen retrieval was done by heat pretreatment at 100°C for 20 min in pH 6 citrate buffer (Roth, Karlsruhe, Germany). After blocking with PBS plus 0.3% Triton (Sigma-Aldrich, St. Louis, MO), 5% BSA (PanReac AppliChem, Darmstadt, Germany), and 5% donkey-serum (Biozol, Eching, Germany) for 30 min, slices were incubated with primary antibodies at 4°C overnight (Supplementary Table S1; Supplementary Data are available online at
RNA and DNA analyses
DNA was either extracted from native porcine gut tissue or the respective decellularized SIS using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). DNA amount was determined using Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen, Carlsbad, CA) and normalized to the dry weight. Qualitative control was done by gel electrophoresis with 2% agarose gel. RNA was extracted from tissue samples using the RNeasy Micro Kit plus (Qiagen, Hilden, Germany) with a TissueLyser LT (Qiagen, Hilden, Germany) and following the manufacturer's protocol. iScript™ Reverse Transcription Supermix for real-time quantitative polymerase chain reaction (qPCR) was used to generate cDNA (Bio-Rad, CA). qPCR was carried out using a CFX 96 Real-time system with a C1000 Thermal Cycler (Bio-Rad, CA) and the Sso-Fast Eva Green Supermix (Bio-Rad, CA). The following reaction condition was chosen: 40 cycles of 95°C 10 s, 60°C 10 s, 72°C 30 s. Exon-spanning primer pairs were either selected from previous literature1,9,23,24 or self-designed using NCBI-Primer designing tool and manufactured by a supplier (Eurofins Genomics, Ebersberg, Germany) (Supplementary Table S2).
Transport studies
For evaluation of the epithelial transport properties, several standard reference substances were tested. All transport studies were performed at 37°C under static conditions, on an OS and in a flow-through bioreactor. Fluorescein sodium salt (Sigma-Aldrich, St. Louis, MO) as a low permeable substance was used to determine paracellular transport, propranolol hydrochloride (Sigma-Aldrich, St. Louis, MO) as a high-permeable substance for transcellular transport, and rhodamin123 (Sigma-Aldrich, St. Louis, MO) as substrate for the efflux-transporter p-glycoprotein (Mdr1). Reference substances were added in a volume of 300 μL basal medium to the apical compartment using following concentrations: 400 μg/mL fluorescein, 30 μg/mL propranolol, and 5 μg/mL rhodamin123. Samples (100 μL) of the basolateral lumen (total 900 μL) were taken between 0 and 120 min every 15 min and replaced with fresh medium. Quantitative analyses of fluorescein and rhodamin123 was determined by measurement of the fluorescence intensity using a microplate reader (Tecan; Infinite M200, Maennedorf, Switzerland). Propranolol concentration was analyzed by HPLC-MS/MS (Shimadzu Nexera LC-30AD, CTO-20AC with FCV-12AH, 8030 Plus; Shimadzu Corporation, Kyoto, Japan). The apparent permeability coefficient (Papp-value) was calculated for every substance. 25
Nanoparticle permeation studies
Poly(
Statistical analyses
Statistical analysis were performed with unpaired t-test with Welch's correction or with one-way ANOVA, Tukey's multiple comparison test. All statistical tests were done by GraphPad Prism 6 Software (GraphPad Software, Inc.). Results are given as mean ± standard deviation. The level of statistical significance is indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. A p-value between 0.05 and 0.15 was additionally considered as moderate evidence for statistical significance and biological relevance (0.05 < #p < 0.15).
Results
The central goal of our study was to provide an improved intestinal test system, which includes the essential requirements to reflect the in vivo physiology. This comprises different primary epithelial cell types of the gut, subepithelial mesenchymal cells, extracellular matrix support, and physiological shear stress (Fig. 1). In the following we introduce the different components within individual sections.
Preparation of a biological scaffold (SIS) for gut tissue engineering
The intestinal model developed in this study includes a decellularized biological scaffold generated from porcine jejunum (Fig. 2). After the mucosa was mechanically removed, decellularization and gamma-sterilization, the obtained matrix macroscopically appeared completely whitish (Fig. 2B). Masson trichrome staining and SEM analysis visualized the largely preserved collagen fibers of the subepithelial connective tissue (Fig. 2C, D, and H). Feulgen staining and DNA isolation confirmed that no significant DNA residuals could be detected after the decellularization process (Fig. 2E, F, and H). This was also confirmed in a quantitative analysis by PicoGreen® dsDNA assay (mean 219 ng DNA per mg dry weight, n = 3).
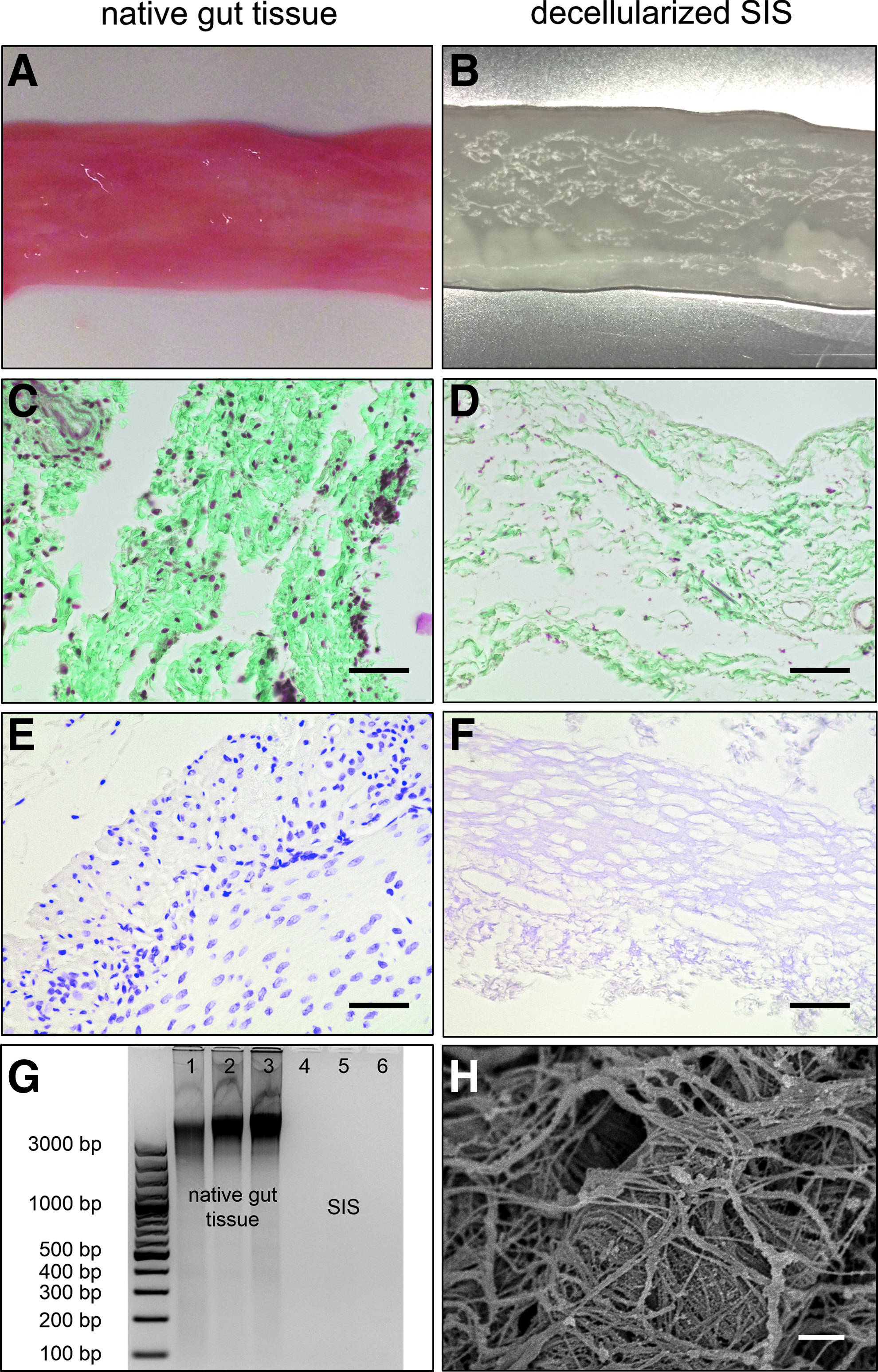
Decellularized biological matrix (SIS) used for intestinal model. Visual control of porcine tissue before and after the decellularization process
Propagation of primary human intestinal epithelial cells
Human crypt-derived primary IECs, isolated from jejunum tissue were embedded in Matrigel allowing formation of intestinal organoid structures according to the protocol published by Sato et al. (Fig. 3A). 10 These organoids contained the main differentiated cell types and undifferentiated, proliferating stem and progenitor cells in crypt niches indicated by immunostaining for the proliferation marker Ki67 (Fig. 3B). To provide enrichment of these proliferating cells, organoids were kept relatively small (size ∼150–200 μm) and passaged every 5–7 days. For quantitative analysis cells were labeled with EdU, which is incorporated into the DNA during cell division. Subsequent flow cytometry analysis demonstrated a large fraction of ∼29.5% EdU-positive cells (n = 4) (Fig. 2C).
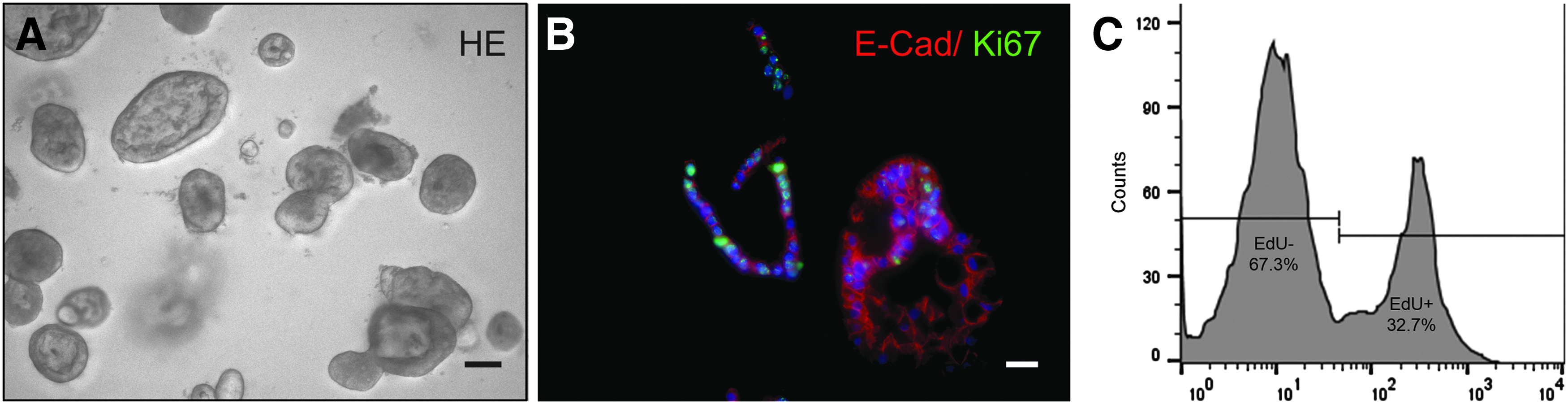
Propagation of intestinal organoids. Intestinal epithelial cells growing as organoids embedded in Matrigel®
Establishment of an intestinal Transwell-like in vitro model
IECs, propagated as organoids, were prepared as single cells and seeded on the decellularized SIS matrix up to 14 days under static culture conditions. Furthermore, we analyzed the biological effects of intestinal subepithelial fibroblasts on barrier integrity. HE-staining of the monoculture (Fig. 4A) demonstrated a confluent monolayer formation on top of the matrix after 5–7 days, which remained largely unchanged in terms of TEER and paracellular FITC-dextran transport during extended culture periods. Interestingly, coculture with fibroblasts led to a more heterogeneous monolayer with partly high prismatic cells and luminal cystic structures within the epithelium (Fig. 4B; black asterisk). The vimentin-immunostained fibroblasts were located directly underneath the E-cadherin-positive epithelium (Fig. 4C). The TEER-measurement revealed relatively low (compared to Caco-2 model) and no significantly different values between mono- and coculture (both >40 Ωcm2). The FITC-dextran permeability was much more robust (i.e., ∼1% vs. 4% after 30 min) in the coculture compared to the monoculture (Fig. 4D, E) confirmed by significant differences between the variances of the two groups. Additionally, gene-expression analysis showed no significant differences neither in the goblet cell-specific Mucin 2 nor the tight junction-specific gene Claudin 4 between the mono- and the coculture (Fig. 4F). In contrast to organoids treated with DAPT, the Transwell-like cultures showed a fourfold reduction of Mucin 2 expression and 15-fold higher expression of claudin 4.

Establishment of intestinal coculture on decellularized SIS. HE-staining of mono-and coculture shows a tight epithelial layer on top of the biological matrix (
To prove the performance of our Transwell-like system for (pre-)clinical relevant NP uptake studies, we applied PLGA-NPs (1 mg/mL) with an average size of 214 nm over a time course up to 6 h. In contrast to an empty SIS, nanoparticles were first detected basolaterally after 1–2 h followed by a linear transport over time (Fig. 5A). After 6 h we detected about 5.5% transported NPs compared to the apically applied amount. Confocal imaging confirmed the uptake of the fluorescence-labeled NPs (green) into the cytosol of the IECs (Fig. 5B). To verify the actual particle transport through the cells and membrane, we performed SEM imaging on representative samples taken from the basolateral compartment after 6 h and detected particles (Fig. 5C).
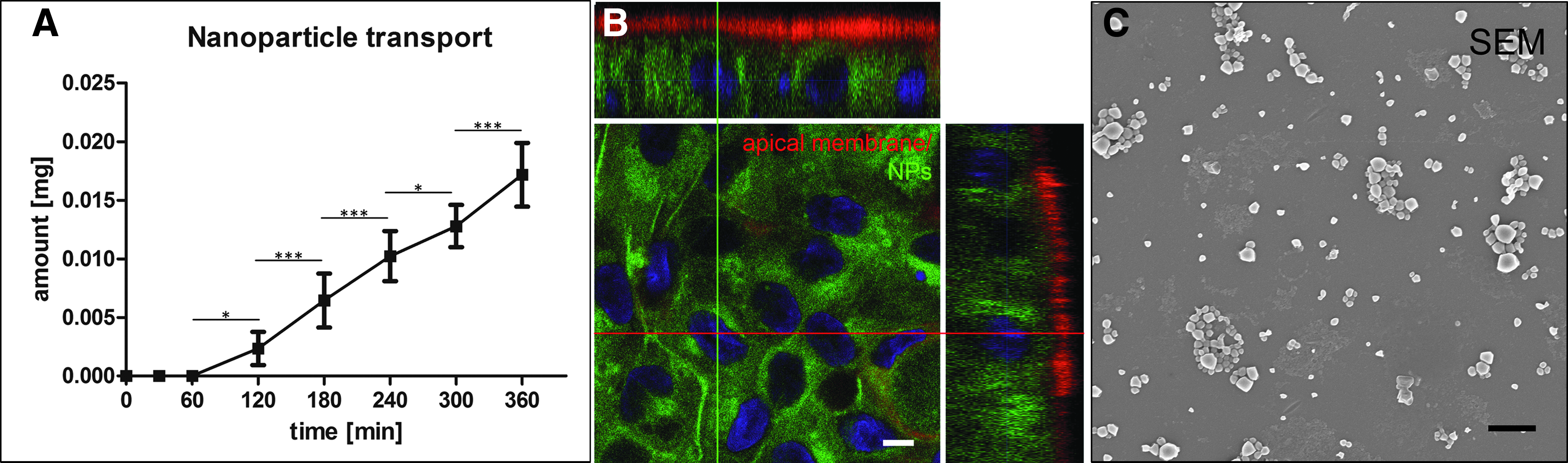
Functional NP uptake studies. Linear transport of PLGA-NPs (1 mg/mL) was tested over a time period of 6 h and showed significant increase of particle transport after 120 min (
Influence of controlled dynamic culture conditions on intestinal Transwell-like culture
In vivo, intestinal cells are continuously exposed to shear stress and dynamic flow conditions. Therefore, we compared static, OS, and dynamic-bioreactor (BR) conditions. For the static and OS setting we used cell crowns allowing fixation of membranes and culture in a standard 24-well format (Fig. 6A). For mechanical stimulation, scaffolds with cells were either placed on an OS (77 rpm) or within a bioreactor system (BR) allowing to apply defined, consistent shear stress to the tissue (Fig. 6B). For the BR, we chose a flow rate of 3.8 mL/min, which has previously been proven as beneficial on intestinal differentiation. 11 Computational modeling demonstrated an average shear stress distribution of 7.1 × 10−3 ± 7.3 × 10−4 dyne/cm2 for the shaker (Fig. 6C) and 6.2 × 10−3 ± 1.0 × 10−3 dyne/cm2 for the bioreactor (Fig. 6D) across the scaffold surface. These values correspond well to mechanical stimulation found in vivo for the intestine. 12
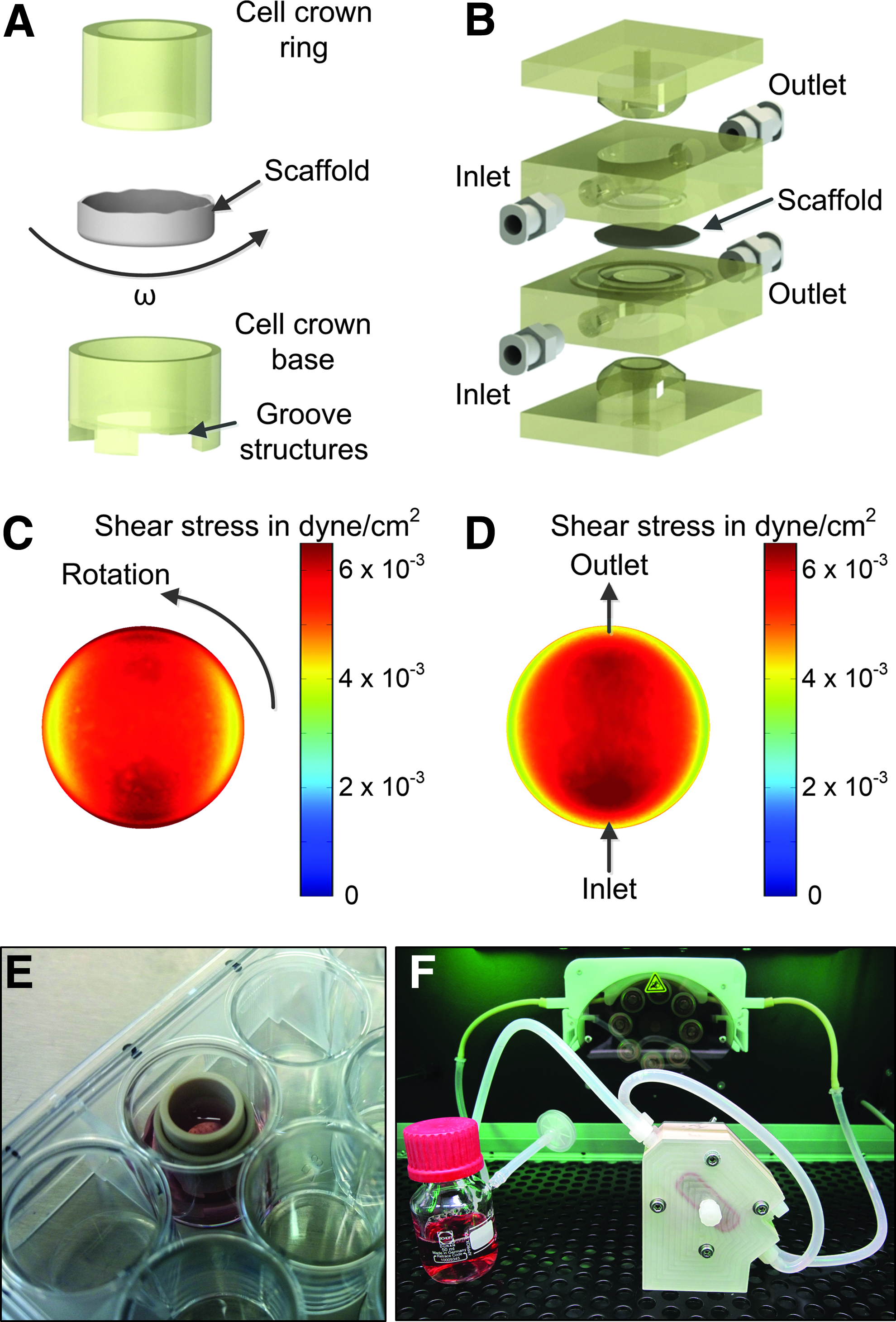
Culture devices and fluid dynamic characterization. The cell crown system supported usage of a biological scaffold in a 24-well
On a histological level, primary IECs cultured in the bioreactor (Fig. 7C) showed a more physiological high prismatic cell morphology (Fig. 7A–C). As mentioned, in the static culture (Fig. 7A) fibroblasts remained directly under the epithelium; in contrast, on the shaker and bioreactor (Fig. 7B, C) cells also migrated into the matrix. On a transcriptomic level, applied culture conditions induced a fivefold downregulation of Mucin 2 compared to the organoids, while Claudin 4 expression was upregulated 6–15-fold, respectively (Fig. 7D). However, expression of the metabolic enzyme CYP3A4 and the important efflux transporter Mdr1 were significantly increased more than threefold under the bioreactor conditions only (Fig. 7E, F).
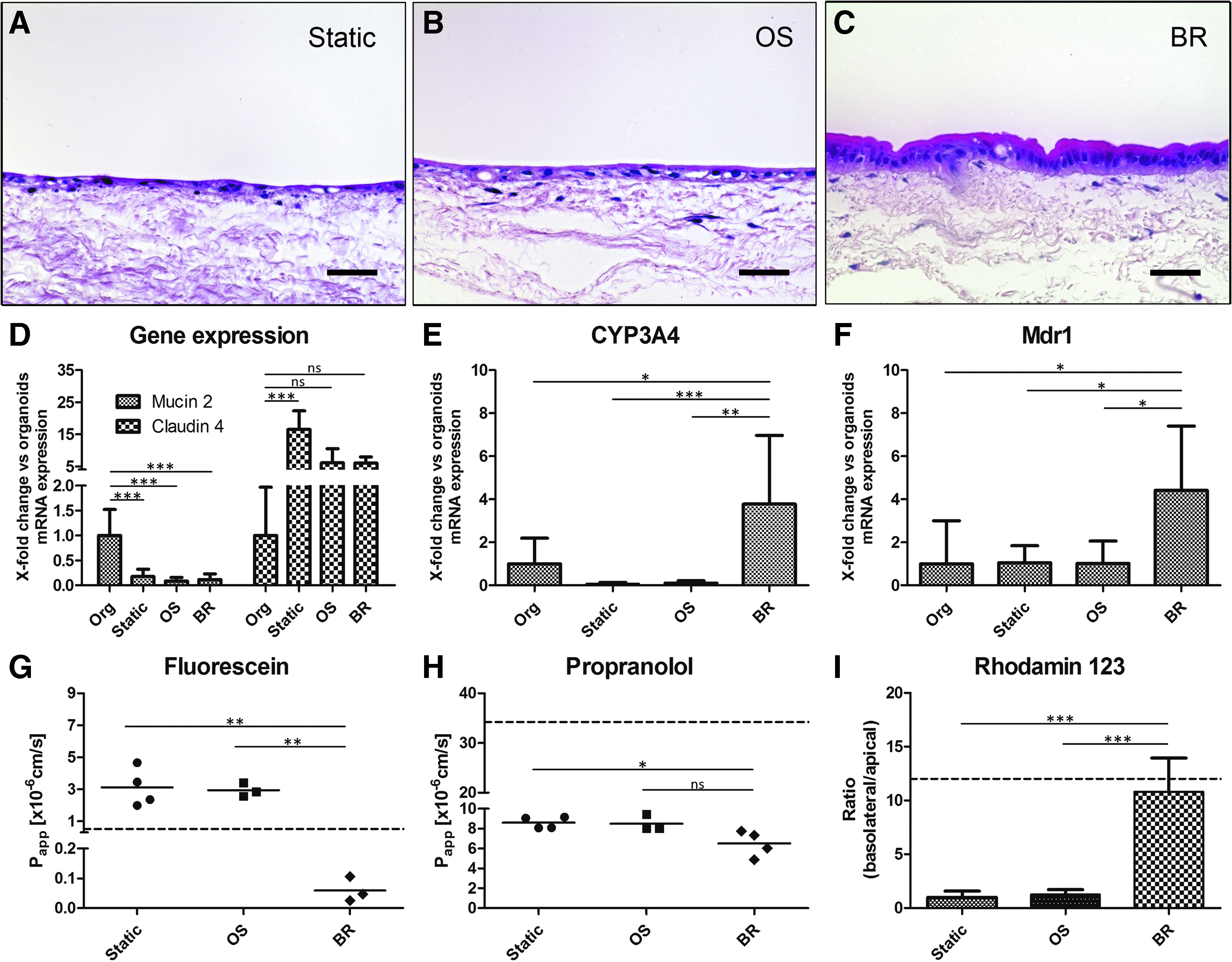
Analysis of static compared to dynamic culture conditions on a shaker (OS) and in a bioreactor (BR). H&E staining of static culture
For a functional proof of transport activity we furthermore applied typical reference substances with known transport properties. Fluorescein as a low permeable substance showed 3 × 10−6 cm/s higher Papp-values in the static and OS compared to the BR conditions with less than 1 × 10−7 cm/s, which lies below the Caco-2-model (dotted line) (Fig. 7G). Similar effects were observed for propranolol with Papp-values of about 6.5 × 10−6 cm/s, which is about 2 × 10−6 lower compared to the other conditions and more than five times lower than in the Caco-2-model (Fig. 7H). Instead, the ratio between basolateral-apical (b/a) and apical-basolateral (a/b) rhodamin123 transport revealed a dramatically increased efflux transporter activity of more than 10-fold under the bioreactor conditions, which is comparable to the Caco-2-model (Fig. 7I).
Immunohistological characterization of the intestinal coculture model under optimized bioreactor conditions
In contrast to commonly used cell line models, the here developed primary model includes the main characteristic cell types also found in the small intestine in vivo as shown in a detailed histological analysis. Thus, the epithelial monolayer was not only positively immunostained for the general epithelial markers cytokeratin 18, pan-cytokeratin, and E-cadherin (Fig. 8A–F), but also for Mucin 1 and Mucin 2 (Fig. 8C, F; green: mucus-producing goblet cells) and Villin (Fig. 8B; green: absorptive enterocytes), which together represented the majority of cells in the monolayer. Furthermore, we detected also hormone-producing enteroendocrine cells immunopositive for chromogranin A (Fig. 8D; green) and paneth cells secreting lysozyme (Fig. 8E; green). These cells were identified as single cells equally distributed over the whole culture. SEM and TEM visualized a cell surface completely covered with microvilli (Fig. 8G, H). The ultrastructure analysis further confirmed tight-junction complexes and desmosomes at the cell–cell-border (Fig. 8H).

Histological characterization of intestinal coculture model under bioreactor flow conditions. Immunohistological stainings of CK18 (Cytokeratin 18), pan CK, and E-Cad (E-cadherin) showed IECs
Discussion
Cell-line-based in vitro models such as Caco-2 are routinely used to investigate toxicity and uptake of orally delivered drugs.25,27,28 Despite many advantages, these models lack important gut characteristics including specific transporter activity and mucus production. 29 Therefore, previous research focused on developing primary human 3D models of the human small intestine, eventually leading to the primary gut organoid technique. 10 However, standardized drug transport studies and infection studies are not feasible with organoid models.30,31 Meanwhile, a few primary Transwell-like barrier models with two separated compartments have been developed using various cell sources from ileum, rectum, 1 or other parts of small intestine 32 using fetal, 33 postmortem tissue (MatTek) or unknown donor origin. Like Caco-2 assays these models also based on porous PET membranes as scaffold,1,32–34 which contain unequally distributed pores leading to impermeable areas and thus a significant artificial barrier for specific test formulations. This has been tested in a direct comparison of paracellular and transcellular drug transport after application on empty and Caco-2-seeded PET membranes or decellularized SIS. 13 Similarly, PLGA nanoparticles designed for our study have been used in a (pre-)clinical trial and were transported much faster in our model compared to related studies using PET membranes.35,36 From literature it is known, that only 0.4–0.6%, of the encapsulated coumarin, is released over 5–48 h.37,38 That is why the percentage of false positive results within the uptake studies is lower than 1%. In addition to altered barrier properties, our decellularized porcine gut matrix (SIS) has been shown to promote (intestinal) cell differentiation,11,15,16,39 allows migration of coculture cells (e.g., fibroblasts) into the matrix, and needs no artificial coating with additional ECM components. In a previous more detailed characterization the matrix revealed a molecular structure mainly consisting of collagen type I fibers and smaller amounts of collagen type III, IV, and VI in addition to glycoproteins elastin, laminin, and fibronectin. 40 Cocultured fibroblasts can secrete important factors modulating epithelial cell behavior and signaling pathways such as Wnt,14,40 which could explain the more robust monolayer integrity observed in FITC-dextran transport. In particular, precise determination of the total Wnt-activity might help to fine-tune in vivo-like conditions in the future. 41
TEER measurement provides a noninvasive method to ensure sufficient barrier integrity of the models before any experimental procedure. 42 TEER-values measured in our model are in average about 40 Ωcm2, which is far below the Caco-2 model but in line with Ussing chamber TEER measurements using native human small intestinal tissue and previously published primary intestinal models.7,8,32,33 However, two other primary models also show higher TEER-values ranging between 130 and 900 Ωcm2, which could be caused by different donor tissues and culture protocols.1,34
In our study, we also investigated the influence of mechanical stimuli, which have been shown to influence cell morphology and differentiation of cells.11,12,43–45 While the bioreactor applies a very good controlled and defined shear stress to the cells, inaccurate regulation, unequal movement, and waves can interfere with the system. The applied dynamic culture provided an average shear stress of 0.005 dyne/cm2 to the cells, forces, which are in the range of physiological relevant values of about 0.002–0.08 dyne/cm2 within the human intestine. 12 Despite the differences in viscosity of medium compared to the digested food, we could demonstrate biologically relevant effects of this approach. However, only the perfusion bioreactor lead to more physiological high-prismatic morphology, and increased expression of most important metabolic enzyme CYP3A4 and efflux transport Mdr1. 46 Furthermore, transport of low-permeable fluorescein 47 and high-permeable propranolol 33 was slower in the bioreactor setting and efflux transport of rhodamin123 increased comparable to values observed in the Caco-2-model. 48 In this respect, further functional validation using well-characterized reference compounds according to the FDA recommendations would be of interest. 49
In contrast to cell line models, our model comprises the cellular complexity of the different cell types found in vivo. In particular, mucus-producing goblet cells play an important role as the mucus layer poses an additional physical barrier and the first line of defence against pathogens. 50 Interestingly, chromogranin-producing enteroendocrine cells and lysozyme-secreting paneth cells can be also be found within the model. Further studies are necessary to fine balance the amount of different cell types within the gut epithelium in vitro.
In conclusion, we developed a human primary in vitro model of the small intestine representing the main physiological characteristics found in vivo. It might therefore be a promising tool for more predictive preclinical testings with pharmaceutical substances, probiotic active organisms, or human pathogenic germs. Finally, such a model could eventually help to reduce animal experimentations.
Footnotes
Acknowledgments
This study was funded by the BMBF (PeTrA, 13N11457). Prof. Dr. Georg Krohne (Division of Electron Microscopy; Biocenter of the University of Würzburg) kindly performed electron microscopy. We thank Mona Alzheimer (Research Center for Infectious Diseases, University of Würzburg) for proofreading the article.
Disclosure Statement
The authors disclose any commercial association that might create a conflict of interest in connection with the submitted article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.