
Abstract
Preplating, a technique used to separate rapidly adherent fibroblasts from the less-adherent progenitor cells, has been used successfully to isolate skeletal muscle-derived stem cells. The objective of this study was to determine if preplating could also be applied to enrich tendon-derived progenitor cells (TDPCs) before monolayer expansion. Cell suspensions obtained by collagenase digestion of equine lateral digital extensor tendon were serially transferred into adherent plates every 12 h for 4 days. TDPC fractions obtained from initial (TPP0), third (TPP3), and seventh (TPP7) preplate were passaged twice and used for subsequent analyses. Growth/proliferation and basal tenogenic gene expression of the three TDPC fractions were largely similar. Preplating and subsequent monolayer expansion did not alter the immunophenotype (CD29+, CD44+, CD90+, and CD45−) and trilineage differentiation capacity of TDPC fractions. Overall, TDPCs were robustly osteogenic, but exhibited comparatively weak adipogenic and chondrogenic capacities. These outcomes indicate that preplating does not enrich for tendon-derived progenitors during in vitro culture, and “whole tendon digest”-derived cells are as appropriate for cell-based therapies.
Introduction
T
Although the benefits of MSC-based therapies for tendinitis and other musculoskeletal injuries have been well established, their clinical use depends on identifying a suitable cell source, isolating a homogenous progenitor population, and efficiently expanding the progenitor cells up to clinically applicable numbers, while maintaining their therapeutic efficacy. Currently, a number of techniques to enrich MSCs from primary cell isolates have been developed. These include cell separation based on size and shape,8,9 expansion from initial low-density plating, 10 differential attachment to plastic or specific substrates11,12 and/or presence of specific cell surface markers.13–15
Tendon-derived progenitor cells (TDPCs) constitute a promising resource for tendon regeneration.16,17 Several recent studies have characterized TDPCs on the basis of previously established MSC cell surface markers.18–20 In all these studies, low-density plating and subsequent colony formation were used to isolate TDPCs. Immunophenotyping TDPCs and selective enrichment using MSC markers have proved to be problematical as markers exclusive to the tenogenic phenotype are limited.21,22 Currently, isolation of homogeneous TDPCs relies on serial passage subculture, to enrich for rapidly and persistently proliferative stem cells from initial heterologous tendon digest populations. Time-dependent adherence, or “preplating,” is a marker-independent isolation method, which segregates rapidly adherent fibroblasts from the less-adherent progenitor cells. This approach has been successfully used to isolate stem cells from skeletal and cardiac muscle tissues.23–25 Muscle and tendon tissues are both mesenchymally derived tissues with relatively low cell densities and a hierarchically arranged, predominantly fibrillar extracellular matrix.26,27 Furthermore, the cellular compartments of both tissues contain heterogeneous populations with very few progenitor cells.18,23 The objective of this study was to determine if preplating enriches TDPCs from tendon digests before monolayer expansion. To test this hypothesis, preplate-selected TDPC subpopulations were characterized by immunophenotyping and standard trilineage differentiation assays.
Materials and Methods
TDPC isolation and preplating
All procedures were approved by the University of Illinois Institutional Animal Care and Use Committee. Hindlimb lateral digital extensor (LDE) tendon was harvested from four young adult horses (2–4 years of age) euthanized for reasons unrelated to musculoskeletal disease. A 1–2 cm length of LDE tendon specimen was diced into 0.25-cm3 pieces and digested in 0.2% collagenase (Worthington) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2% fetal bovine serum (Gemini Biomedicals) at 37°C for 16 h.28,29 The cells were isolated by filtration and centrifugation, and the cells were seeded at 500 cells/cm2 in monolayer cultures in high-glucose DMEM supplemented with 20% fetal bovine serum, 37.5 μg/mL of ascorbic acid, 300 μg of
Monolayer expansion and cell proliferation
TDPC fractions isolated by preplating were trypsinized at ∼80% confluency. Viability was determined by trypan blue exclusion and the resultant cells were seeded at 1 × 104 cells/cm2 and expanded in monolayer cultures in high-glucose DMEM supplemented with 10% fetal bovine serum, 37.5 μg/mL of ascorbic acid, 300 μg of
Flow cytometry and immunophenotyping
Third passage TDPCs from TPP0, TPP3, and TPP7 were used for single-color flow cytometry. Aliquots of 1 × 106 TDPCs were fixed in 4% paraformaldehyde and then blocked with 1% bovine serum albumin in high-glucose DMEM for 20 min. Cells were washed in phosphate-buffered saline (PBS) and resuspended in either fluorescent-conjugated or -unconjugated primary antibodies and incubated at 4°C for 30 min. Flow cytometry was used to evaluate the MSC immunophenotype (CD29, CD44, and CD90) during monolayer expansion, following previously published protocols. 30 CD45 was included as a negative control for hematopoietic progenitors. The following antibodies were used according to the manufacturers’ recommendations: anti-human conjugated anti-CD29-Alexa 488 (BioLegend); anti-horse conjugated anti-CD44-RPE (AbD Serotec; Bio-Rad); anti-horse nonconjugated anti-CD90-Alexa 647 (Accurate Chemical and Scientific Corporation), and anti-human conjugated anti-CD45-Alexa 488 (AbD Serotec; Bio-Rad). 30 The following filters were used in a flow cytometry analyzer (Accuri C6; BD Biosciences) to isolate the emission wavelength of the conjugated fluorochromes: FL-1 (510 and 545 nm wavelengths of light) for CD29 (519 nm emission) and CD45 (519 nm emission), FL-2 (560–580 nm wavelength) for CD44 (578 nm emission), and FL-4 (665–695 nm wavelength) for CD90 (668 nm emission). After the emission analysis on “FCS express (Flow Research Edition),” data were expressed as “percentage of deviation from the control antibody groups.” Bone marrow-derived MSCs were used as reference controls. Cells in the absence of antibody and presence of secondary antibody only were used as controls. A threshold gating out at least 95.5% of control cells was used.
Tenogenic gene expression
Three million third passage TDPCs from TPP0, TPP3, and TPP7 were stored for RNA isolation before trilineage differentiation experiments. Basal expression of tenogenic genes, scleraxis (Scx), and tenomodulin (Tnmd) was assessed by quantitative polymerase chain reaction (PCR), as detailed below.
Trilineage differentiation and phenotypic staining
Osteogenic culture
Passage 3 TDPCs from TPP0, TPP3, and TPP7 were plated at 5 × 103 cells/cm2 in six-well plates and cultured in complete DMEM until they reached 80% confluence. Complete DMEM was then substituted with osteogenic media (complete DMEM supplemented with 10 mM β glyceraldehyde-3-phosphate, 50 μg/mL ascorbic acid, 100 ηM dexamethasone). The media were replaced every 3 days.11,20 The cultures were maintained for 14 days.
Alizarin Red staining was used to assess mineralized matrix deposition. The cell–matrix layer was washed with PBS, fixed with 70% ethanol, and stained with 2% Alizarin Red stain for 10 min. Low-magnification (10 × ) images were obtained before osteogenic differentiation and at days 7 and 14 of osteogenic culture. Upregulation of osteogenic genes was also assessed by quantitative PCR, as detailed below.
Adipogenic culture
Passage 3 TDPCs from TPP0, TPP3, and TPP7 were cultured in six-well plates with complete DMEM until they reached 80% confluence. Complete DMEM was then substituted with adipogenic media (high-glucose DMEM containing 10% rabbit serum, 100 U of sodium penicillin/mL, and 100 μg of streptomycin sulfate/mL and supplemented with 1 mM dexamethasone, 100 mM indomethacin, 10 mg/mL insulin, and 500 mM isobutylmethylxanthine).11,20 Media were replaced every 2 days. These cultures were maintained for 10 days.
Oil-Red-O staining of monolayers was used to detect intracellular lipid accumulation. Cell monolayers were washed with PBS, fixed with 70% ethanol, and stained with 0.3% Oil-Red-O stain for 1 h. Low- (10 ×) and high-magnification (50 ×) images were obtained before adipogenic differentiation and at days 3 and 10 of adipogenic culture. Upregulation of adipogenic genes was also assessed by quantitative PCR, as detailed below.
Chondrogenic culture
Pellet cultures were established in microcentrifuge tubes from passage 3 TDPCs by resuspending 5 × 105 cells/mL in chondrogenic media (high-glucose DMEM supplemented with 100 ηM dexamethasone, 25 μg/mL ascorbic acid, 10 ηg/mL TGF-β1, and 1% ITS media supplement) and pelleting 500 μL aliquots of the cell suspensions at 400 rcf.11,20,30 Chondrogenic cultures were maintained for 20 days. The media were replaced every 3 days.
Representative pellet sections were stained with toluidine blue to assess sulfated glycosaminoglycan (sGAG) deposition. 30 After 20 days, cell pellets were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. Six-micron-thick sections were stained with toluidine blue. High-magnification (50 ×) histological images were acquired with LEICA Q500MC microscope (Leica Cambridge Ltd.). Upregulation of chondrogenic genes was also assessed by quantitative PCR, as detailed below.
RNA isolation and quantitative reverse transcription polymerase chain reaction
Total RNA was isolated using a previously described protocol.28,30 The samples were homogenized in a guanidinium thiocyanate-phenol-chloroform solution reagent (TRIzol; Invitrogen) according to manufacturer's suggested protocol. RNA isolation from the chondrogenic pellets included the high-salt precipitation variation recommended by the manufacturer, to minimize coprecipitation of proteoglycans.
30
The resultant pellet was purified using RNeasy silica columns that included on-column DNase digestion. One microgram of RNA from each sample was reverse-transcribed (Superscript II; Invitrogen) using oligo(dT) primers. Equine gene-specific primers were designed from published sequences in GenBank, and using ClustalW multiple sequence alignment (available at
Adpn, adiponectin; Agg'n, aggrecan; ALP, alkaline phosphatase; EF1α, elongation factor-1α; FABP4, fatty acid binding protein-4; Osn, osteonectin; Osx, osterix; Scx, scleraxis; Tnmd, tenomodulin.
Statistical analysis
The normality of distribution of quantitative data (relative mRNA expression) was confirmed using the Kolmogorov–Smirnov test using SigmaStat 4 software (Systat Software). Data are expressed as mean ± standard error. One-way ANOVA was used to assess the effect of preplating on cell proliferation and differentiation, in the three fractions of TDPCs (TPP0, TPP3, and TPP7). A p-value of ≤0.05 was considered significant.
Results
Cell culture and proliferation
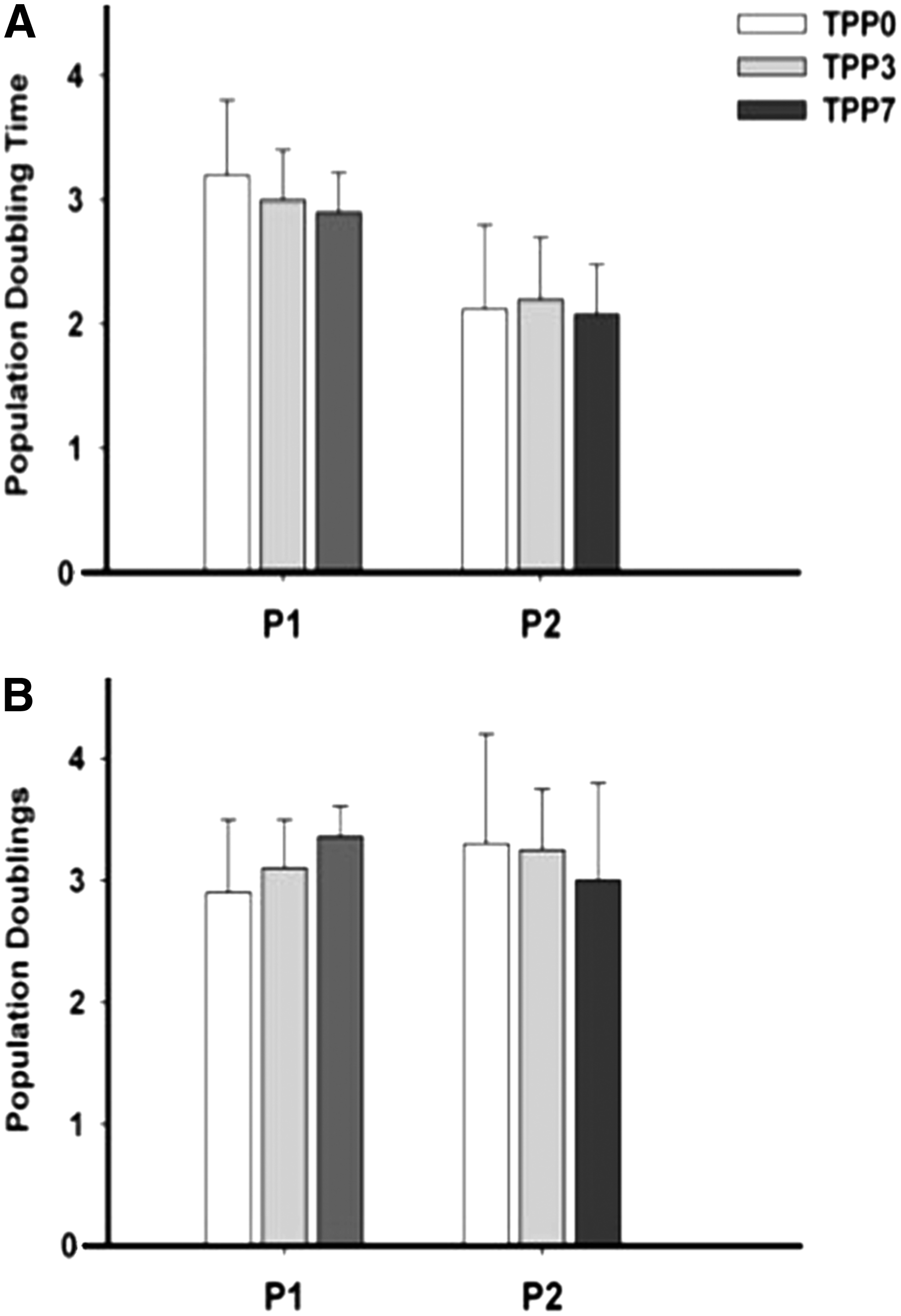
The time from initial plating of each preplated TDPC subpopulation to 80% confluence was significantly less in TPP0 (4 ± 0.9 days; p = 0.034) than in TPP3 (9.8 ± 0.9 days) and TPP7 (12.3 ± 1.2 days). In addition, the more slowly adherent TDPC subpopulations were quiescent for the first 5–6 days after transfer, before cell division and colony formation. Population doubling time and population doublings during P1 and P2 were not significantly different between the TDPC fractions (Fig. 1).
Immunophenotype
More than 90% of cells from all TDPC fractions were immunopositive for CD29, CD 44, and CD90; surface markers that characterize equine MSCs. All fractions of TDPCs were negative for the hematopoietic marker, CD45 (Fig. 2). There were no significant differences (p = 0.7) in the immunophenotypes of TPP0, TPP3, and TPP7.
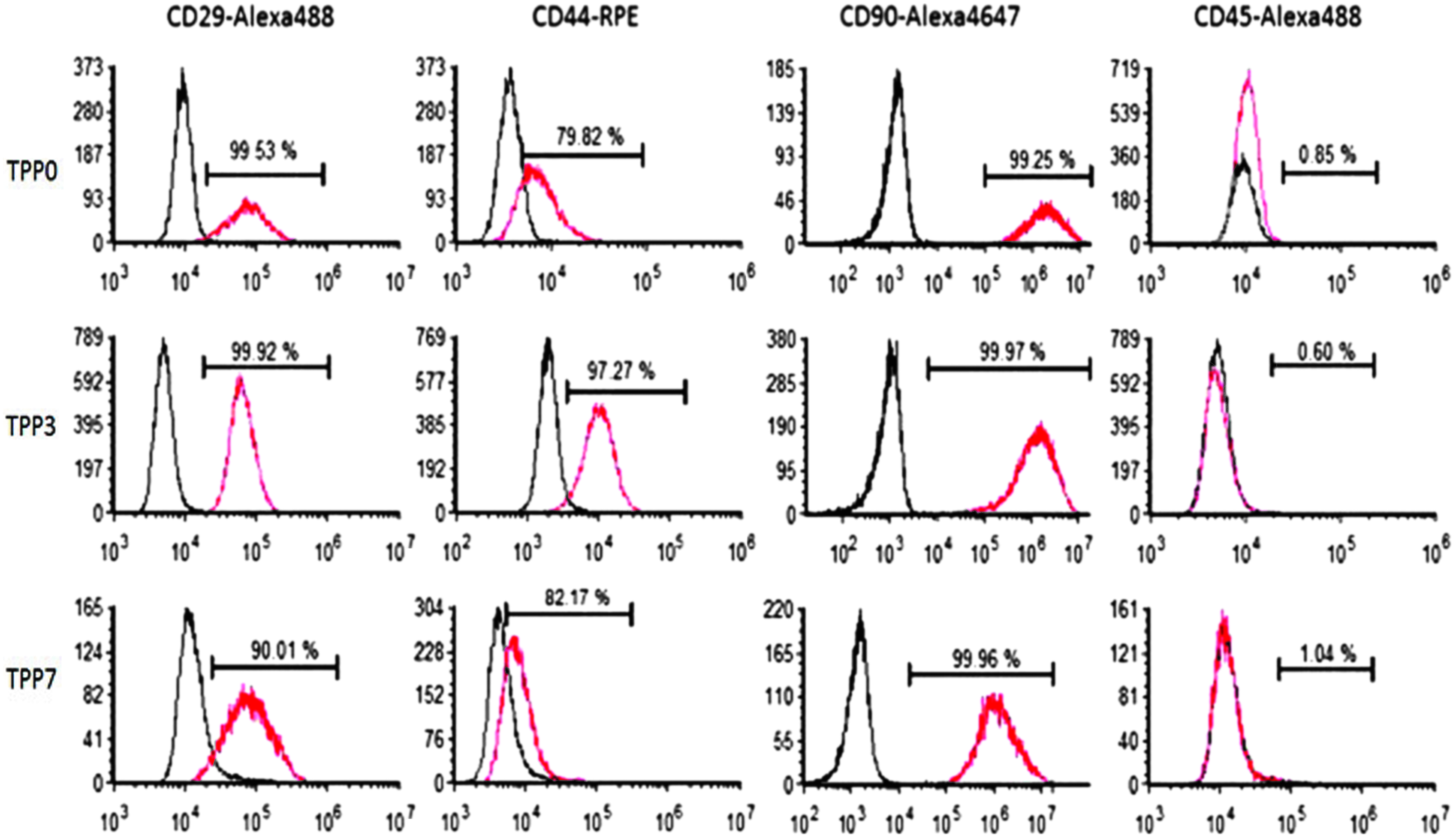
Representative immunophenotypic profile of third-passage TDPC fractions from a single donor. The red profiles indicate the distributions of the epitope-specific antibodies, while the black profiles represent distributions of non-immune control antibodies. Color images available online at
Tenogenic gene expression
The basal mRNA expression of tenogenic markers, Scx and Tnmd, was similar in all TDPC fractions (p > 0.1) (Fig. 3).
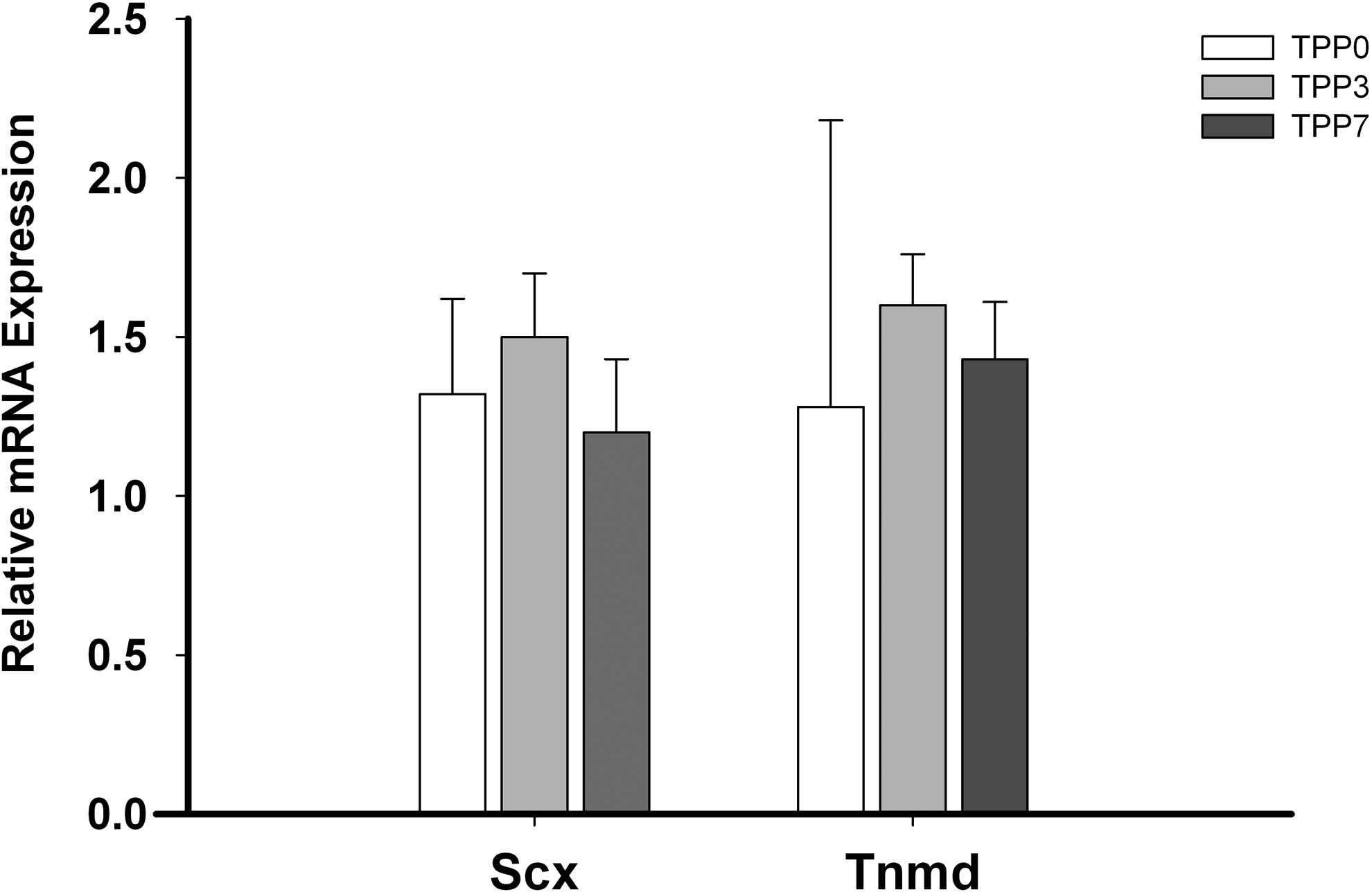
Relative mRNA expression of tenogenic genes Scx and Tnmd in TDPC fractions (mean ± SE; n = 4). Scx, scleraxis; Tnmd, tenomodulin.
Trilineage differentiation
Osteogenesis
Alizarin Red staining of the mineralized matrix in day 7 osteogenic TDPC cultures showed minimal stain uptake in all fractions. By day 14, intense staining of mineralized nodules, indicative of robust osteogenic differentiation, was evident in all TDPC fractions (Fig. 4A). There were no noticeable differences in the intensity of Alizarin Red stain uptake between the TDPC fractions.

Basal mRNA expression of osteogenic transcription factors osterix and Runx2 was similar in all TDPC fractions before osteogenic differentiation. In the osteogenic medium, expression of osterix and Runx2 transcripts was significantly increased (10- to 12-fold; p < 0.05) by day 14 in all TDPC fractions and the expression was not significantly different between them. On day 14 of osteogenic induction, changes in mRNA levels of genes linked to osteogenic phenotype (osterix, osteonectin, alkaline phosphatase) corroborated the Alizarin Red staining outcomes. Expression of osterix, Runx2, osteonectin, and alkaline phosphatase on day 14 of osteogenic culture was not significantly (p = 0.6) different between the TDPC fractions (Fig. 4B).
Adipogenesis
Oil-Red-O staining of adipogenic cultures showed intracellular lipid droplet deposition in all TDPC fractions on day 3, which was higher on day 10 (Fig. 5A).
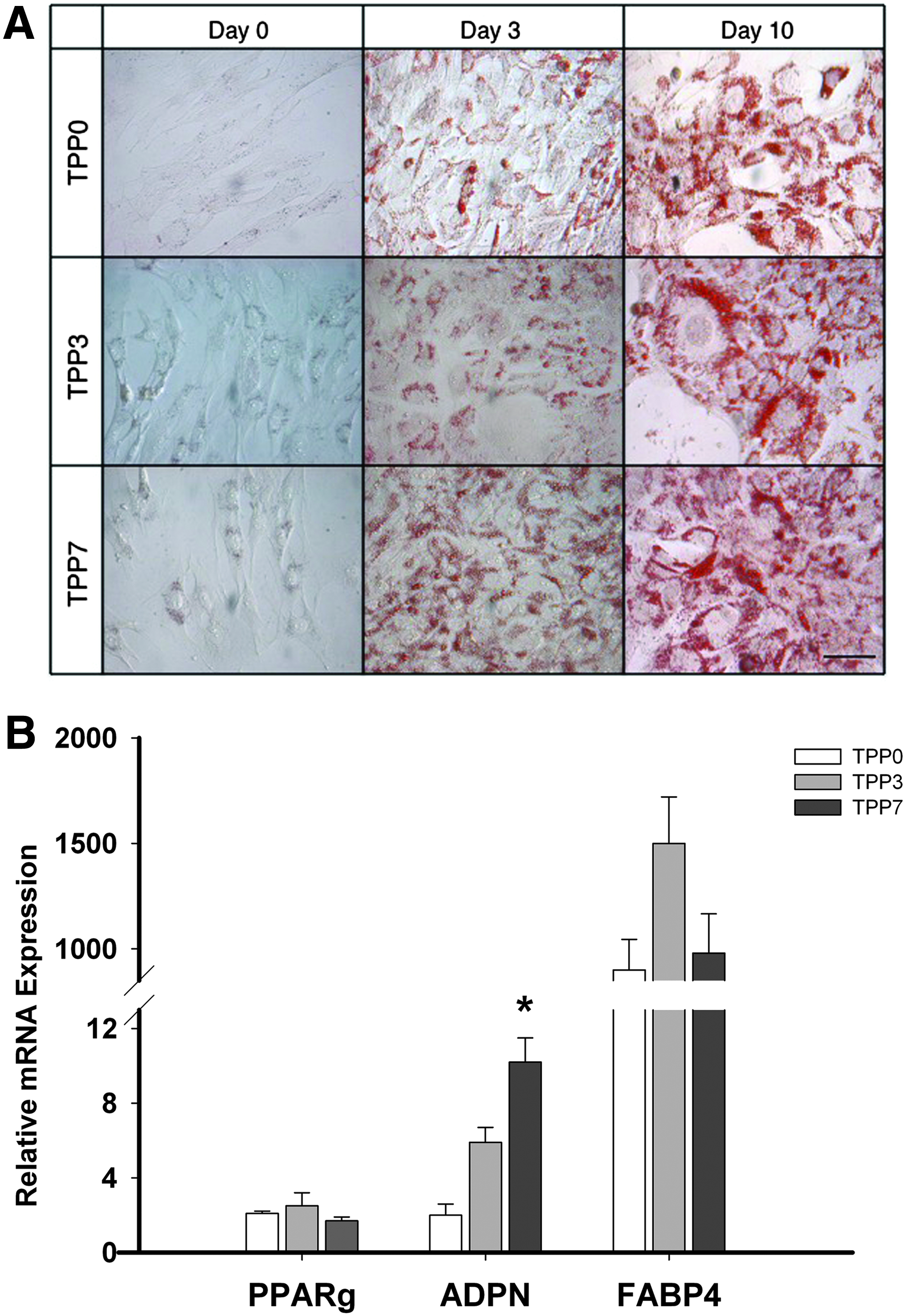
Basal mRNA expression of the adipogenic transcription factor PPARγ was similar in all TDPC fractions. The adipogenic culture medium stimulated a marginal increase (2- to 3-fold) in PPARγ mRNA expression by day 10 in all three TDPC fractions; this increase was not significant (p = 0.4). Changes in the expression of adipogenic genes (PPARγ, FABP4, and adiponectin) were not consistent (Fig. 5B). Expression of adiponectin mRNA on day 10 of adipogenic culture was significantly (p = 0.042) higher in TPP7 compared to TPP0 and TPP3; however, there was no significant difference in the expression of FABP4 in the three TDPC fractions on day 10 of adipogenic culture (Fig. 5B).
Chondrogenesis
Toluidine blue staining intensity of day 20 chondrogenic pellets was minimal in all three TDPC fractions, indicating low sGAG content within the pellets (Fig. 6A).

Basal mRNA expression of Sox-9 was similar in all TDPC fractions. As expected, chondrogenic stimulation significantly upregulated Sox-9 mRNA expression on day 20 compared to baseline, but Sox-9 expression was not significantly (p = 0.31) different between the three TDPC fractions. Chondrogenic matrix gene transcripts (collagen type II and aggrecan) were significantly (p = 0.002) upregulated on day 20 of chondrogenic stimulation compared to baseline in all TDPC fractions (Fig. 6B).
Discussion
This study investigated the value of preplating for TDPC enrichment from tendon digests. We hypothesized that a differential attachment preplating screen would enrich TDPCs in the more slowly adherent fraction(s). Accepting the marked differences in initial seeding densities of the primary cultures, preplating and subsequent monolayer expansion did not alter the growth/proliferation or immunophenotypic characteristics of the TDPC subpopulations. Basal expression of tenogenic markers was similar across the preplate fractions and the osteogenic, adipogenic, and chondrogenic differentiation capacities of the three TDPC fractions were also similar. These outcomes do not support the hypothesis.
Time to confluence was significantly shorter for TPP0 cultures, compared to TPP3 and TPP7. This finding was expected, given that a large proportion of the primary isolates attached during the first preplate. Substantially fewer cells remained unattached after the first serial plating and were accessible for subsequent transfers. The population doubling and population doubling times in subsequent passages were similar across the TDPC fractions, indicating that the “per-cell” proliferative capacities of the subpopulations were similar, consistent with results obtained for muscle-derived stem cells isolated through similar protocols.24,25 Initial quiescence and heterogeneity in colony-forming units noted in this study during in vitro isolation have also been reported in both mouse and human TDPCs 18 and is a common feature of MSCs from other tissue sources.
Allowing for species variations in stem/progenitor cell immunophenotypes, the cell surface marker profiles of equine TDPCs in our study were similar to other studies11,20 and were characteristic of equine MSCs (CD29+, CD44+, CD90+, and CD45−).30,32,33 Preplating and subsequent monolayer expansion did not influence the immunophenotype. As with this study, preplated skeletal muscle-derived stem cell populations did not differ in their MSC surface marker profile; however, preplating did enrich cells expressing markers specific to myogenic stem cells.23,24,34 Scleraxis expression did not differ in the TDPC preplate groups in this study; furthermore, unpublished data from our group indicate that Scx expression in TDPCs and bone marrow-derived MSCs (from the same donors) is equivalent under basal conditions. The transcriptional regulation of tenogenesis is less well characterized than for myogenesis or several other mesenchymal lineages, and Scx expression, by itself, might be inadequate as a selection marker. 35 In support of this possibility, equine TDPCs exhibit several tenogenic characteristics more strongly than their bone marrow-derived counterparts in in vitro models, despite highly similar basal profiles.28,36,37
Basal expression and induction profiles of osteogenic-, adipogenic-, and chondrogenic-lineage genes in response to respective in vitro stimuli were similar in all TDPC fractions. However, the overall osteogenic, adipogenic, and chondrogenic capacities of TDPCs were markedly different. All TDPC fractions underwent robust osteogenic differentiation, evident in both matrix staining and gene expression profiling. The induction of transcriptional adipogenic markers was inconsistent. PPARγ was only modestly upregulated (∼2-fold), while adiponectin induction was the only parameter assayed in this study that was responsive to preplating, exhibiting increased expression in later preplate subpopulations. In marked contrast, there was a significant (over 3 logs) increase in FABP4 expression above baseline expression. These somewhat contradictory results indicate that the indices of adipogenesis addressed in this study are not transcriptionally linked and emphasize the value in evaluating several phenotypic indicators in differentiation studies. Similarly, chondrogenic differentiation of TDPCs was less impressive than seen in other equine progenitor populations.30,38 It is not clear from the outcomes of this study whether the impressive osteogenic capacity of TDPCs is an intrinsic property of these cells or is an aberrant consequence of extensive in vitro proliferation. TDPC-mediated ectopic ossification has been reported during tendon healing,39,40 providing support for the former possibility. The restricted adipogenic and chondrogenic profiles of TDPCs observed in our study are consistent with other studies that characterized TDPCs isolated from healthy tendon by low-density plating 41 and specific substrate adhesion. 11 Fatty and mucoid degeneration,42,43 chondrodysplasia,44,45 and ectopic ossification39,40 have been reported in both naturally occurring and experimental models of tendinitis. These changes reflect aberrant differentiation of progenitor cells and/or transdifferentiation of tenocytes within the tissue. Although the specific mechanism(s) that generates these aberrant phenotypes is undefined, it is likely that the differentiation of TDPCs under inflammatory/healing and homeostatic conditions differ widely. In this respect, TDPCs isolated from healthy tendons with a restricted differentiation potential may be particularly suitable for cell-based therapies, provided a suitable source with minimal donor-site morbidity can be identified.
The developmental origin of TDPCs is largely unknown and research in this area is ongoing. Although markers specific for tenogenic lineage (tenomodulin, scleraxis, mohawk) have been identified, their expression is not limited to tenogenic cells and they are not particularly useful for cell isolation/selection protocols. 21 Other techniques of MSC isolation such as selective substrate adhesion, hypoxic conditions, during in vitro culture also did not enrich TDPCs. 11 Although the results of this study indicate that preplating confers no clear benefit for TDPC enrichment, these outcomes and the results of related studies indicate that “whole tendon digest” cell stocks are of significant therapeutic value for tendon repair.16,29,46
Conclusion
In conclusion, differential adherence preplating did not enrich equine TDPC isolation during in vitro culture and monolayer expansion. Preplating did not alter the in vitro growth/proliferation characteristics of TDPCs. The immunophenotype and trilineage differentiation potential of the three TDPC fractions assessed in this study were similar. Overall, TDPCs had a robust osteogenic capacity and a minimal adipogenic and chondrogenic capacity. The results of this study indicate that whole digest tendon-derived cell stocks are adequate for enrichment of progenitor cells by monolayer expansion.
Footnotes
Acknowledgment
This study was funded by the United States Department of Agriculture's Section 1433 Animal Health and Diseases program.
Disclosure Statement
No competing financial interest exists.