
Abstract
The therapeutic infusion of adipose-derived stromal vascular fraction (SVF) cells for the treatment of multiple diseases, has progressed to numerous human clinical trials; however, the often poor retention of the cells following implantation remains a common drawback of direct cell injection. One solution to cellular retention at the injection site has been the use of biogels to encapsulate cells within a microenvironment before and upon implantation. The current study utilized three-dimensional bioprinting technology to evaluate the ability to form SVF cell-laden spheroids with collagen I as a gel-forming biomatrix. A superhydrophobic surface was created to maintain the bioprinted structures in a spheroid shape. A hydrophilic disc was printed onto the hydrophobic surface to immobilize the spheroids during the gelation process. Conditions for the automated rapid formation of SVF cell-laden spheroids were explored, including time/pressure relationships for spheroid extrusion during bioprinting. The formed spheroids maintain SVF viability in both static culture and dynamic spinner culture. Spheroids also undergo a time-dependent contraction with the retention of angiogenic sprout phenotype over the 14-day culture period. The use of a biphilic surface exhibiting both superhydrophobicity to maintain the spheroid shape and a hydrophilicity to immobilize the spheroid during gel formation produces SVF cell-laden spheroids that can be immediately transplanted for therapeutic applications.
Introduction
T
Previous work toward SVF cell aggregation has included the use of alginate encapsulation to form spheroids 22 and three-dimensional (3D) culture of SVF in collagen gels. 23 Current techniques for the creation of spheroids fall primarily under the nominal categories of cellular spheroids, surface seeded spheroids, and encapsulated spheroids. Cellular spheroids are typically created by hanging drop methods or otherwise rely on the mechanism of cells self-aggregating due to lack of other adhesion points.24,25 This production process takes several days to produce spheroids and suffers from inconsistencies that are heavily magnified by cell proliferation rates.
Surface seeded spheroids are prefabricated with the biopolymer of choice and then incubated with the cells of choice to allow cell migration-driven seeding.26–29 This production strategy has a very broad range of applications so long as the biomaterial of choice can be prefabricated. The drawback of prefabricated spheroids is that the cell seeding is migration/adhesion based and, therefore, is primarily limited to the spheroid surface rather than the entire volume of the spheroid limiting the spheroid loading capacity. The third strategy of encapsulated spheroids revolves around the idea of mixing cells into a suspension and then allowing that suspension to become the scaffold/hydrogel of choice.22,30
Encapsulation-based spheroids have the advantage of being able to utilize the full volume of the spheroids for cell carrying and the ability to be produced in less than a day. This strategy has been very biomaterial specific and, in the case of collagen, frequently required the use of a support material, such as alginate that alters the mechanical properties and by proxy some of the biological properties that made the original biomaterial so appealing. The use of a biphilic surface removes the need for a support material and should be applicable with most hydrogels.
Extrusion 3D printing is a well-established and highly customizable production method within the additive manufacturing umbrella. Within the subfield of bioprinting, extrusion bioprinting has the advantage of being compatible with a wide range of materials in comparison to other additive manufacturing approaches, such as those based off of stereolithographic principles that require specific material properties such as light curability. Extrusion bioprinting was selected as our production approach due to its ability to be used for a wider range of applications in comparison to the other current manufacturing methods such as stereolithography.
We have combined the use of 3D bioprinting technology with a superhydrophobic surface to support the automated production of SVF cell-laden spheroids in collagen I gels. A limiting factor in the utilization of bulk collagen gels is the handling before and during implantation. Collagen I polymerization, from liquid to cross-linked hydrogel, typically occurs within a shape holder, such as a multiwell plate This polymerized construct is subsequently removed and handled manually with sterile equipment. Handling, and any potential subsequent gel damage, is very difficult to regulate. In addition, while collagen gels have some elasticity, in vivo applications generally call for shapes that are irregular, therefore, forcing a bulk gel to be either cut into sections or put under significant deformation to fit the required shape. These issues could be mitigated by unitizing collagen gels into small spheroids. Collagen spheroids would have the benefits of simplified handling in a fluid suspension and allow for precise spatial and dosing control to fit in vivo application requirements in a superior manner compared with cell suspension injections alone.
To produce collagen spheroids, a biphilic surface was used in conjunction with a 3D bioprinter to rapidly produce spheroids of consistent size and shape. The two primary reasons that collagen was selected as the material for printing were because of its well-characterized ability to promote SVF capillary network formation and angiogenesis and its relatively extensive characterization as a bioink for printing.31–37 Indeed SVF has demonstrated angiogenesis in 3D collagen I systems as a means to create prevascularized, implantable constructs. 20 Based upon these results and others, SVF encapsulation in collagen I spheroids was the chosen method for this study.
Methods
Fabrication of a superhydrophobic surface
The superhydrophobic surface was formed on a polystyrene 48-multiwell plates (Corning, Corning, NY) and 35-mm Petri dishes using a two-step aerosol application of NeverWet™ (Rust Oleum, Vernon Hills, IL). The first step was the application of a binder to the surface as a base coat, which air dried at room temperature for at least 1 h. This was followed by the application of a top sheet composed of polydimethylsiloxane modified with hexamethyldisilazane to form the superhydrophobic layer. Superhydrophobic layer thickness was measured to be 0.07 mm. The top sheet subsequently air dried at room temperature for an additional hour. NeverWet has a reported contact angle of 165° and a surface is considered superhydrophobic beyond contact angles of 150°. The contact angle of both water and unpolymerized collagen in solution was measured through a side view photograph and subsequent contact angle measurement in ImageJ.
Creation of hydrophilic spots
Hydrophilic spots on the superhydrophobic surface were created using a 3D bioprinter (Bio-Assembly Tool (BAT) 3-D printer; nScrypt, Inc., Orlando, FL) to extrude Pluronic F-127 (Sigma, St. Louis, MO). For each hydrophilic spot, the BAT extruded a target volume of 2 μL of 3.8% (wt/wt) Pluronic F-127 in 1X phosphate-buffered saline (PBS). With the BAT time–pressure extrusion system, this required 2.5 psi with an exposure time of 100 ms through a 25G needle to create the appropriate extrusion force to dispense the target volume. These spots were then allowed to air dry for 30 min before use.
SVF cells
SVF cells were isolated from rat epididymal fat pads according to our previously published enzyme-based methods.38–40 All animal studies were performed under IACUC (Institutional Animal Care and Use Committee) approval from the University of Louisville. Briefly, fat samples were obtained from rat epididymal fat pads under sterile surgical procedure, minced manually for 3 min, washed with PBS that contained 0.1% bovine serum albumin (BSA), and suspended in 2 mg/mL type IV collagenase (Worthington Biochemical Company, Freehold, NJ). The fat was digested for 35 min at 37°C using an Enviro-Genie (Scientific Industries, Bohemia, NY). SVF cells were separated from adipocytes by centrifugation (350 g for 4 min), after which the supernatant was discarded. The pellet was washed twice with PBS, filtered through a 250 μm filter, and resuspended in endothelial cell media to be used immediately for spheroid bioprinting.
SVF cell-laden collagen spheroid fabrication
Freshly isolated SVF was suspended in unpolymerized rat tail collagen I mixed with 1X Dulbecco's modified Eagle's medium (DMEM) (Sigma) titered to a final pH of 7.4 to create a mixture of 3 mg/mL collagen containing 1.6 × 105 SVF cells/mL of final solution. This mixture was kept at 4°C until printing and a refrigerant system on the bioprinter was used to maintain an equivalent temperature throughout the printing process to prevent gel polymerization. The SVF collagen solution was transferred to a 3-mL printing syringe (EFD, Nordson, Westlake, OH) and placed in a 3D Bioprinter (nScrypt, Inc., Orlando, FL). The initial printing conditions were based on our previously published data for continuous cylinder printing.21,32,41,42 Once spheroids were printed, they were incubated at 37°C in a tissue culture incubator (5% CO2) for 10 min to initiate collagen gel polymerization.
Spheroid culture methods
Following fabrication, spheroids were transferred to a spinner flask (125-mL MagnaFlex Microcarrier Spinner Flask; Wheaton Industries, Millville, NJ) for cell suspension culture containing endothelial cell growth media 43 composed of DMEM, 10% FBS, 5 mM HEPES buffer, 2 mM L-glutamine, endothelial growth supplement containing heparin, penicillin (45 U/mL), and streptomycin (45 μg/mL). The spinner flask was placed onto a magnetic stirrer platform (MCS 104-L Biological Stirrer; Techne, Inc., Burlington, NJ) to provide continuous suspension of the spheroids, and the magnetic impeller speed was set to 40 rpm throughout the culture period (14 days). Spheroids required for analysis were removed through a 50-mL serological pipette. For spheroid contraction studies, individual spheroids were plated in separate wells of a 96-well ultra-low attachment plate (Corning, Corning, NY).
Spheroid size measurements
Spheroid size measurements were calculated from images obtained through light microscopy (CKX41; Olympus, Tokyo, Japan) of spheroids cultured in 96-well plates. Diameter measurements of these 4X images were obtained using ImageJ software.
Live/dead assay
SVF viability was assessed using live/dead fluorescent stains (the Live/Dead Viability/Toxicity Kit; Life Technologies, Inc., Carlsbad, CA). Spheroids were washed with PBS, incubated with 10 μM calcein AM (live) and 10 μm ethidium homodimer-1 (dead) at room temperature for 45 min and imaged through epifluorescent microscopy (IX71; Olympus). Images were captured at 4 × magnification. To quantify the viability of cells in the spheroids produced, isolated SVF cell viability was measured using a NucleoCounter (ChemoMetec, Allerod, Denmark) and known amounts of these cells were used to create live and dead fluorescent standard curves in a Cytation 5 Imaging Multi-Mode Reader (BioTek, Winooski, VT). Based on the developed standard curves, viability was measured on days 0, 2, 6, 9, and 13. To supplement the live/dead kit, the distribution of cells was assessed with a Hoechst 33258 bis-benzimide nuclear stain (Anaspec, CA) following 15 min of 0.1% Triton X-100 (Sigma) permeabilization.
Confocal microscopy
For in vitro vascularization analysis, spheroid samples were imaged using an MPE FluoView1000 confocal microscope utilizing a 10X water immersion objective (Olympus). Confocal image stacks were reconstructed in AMIRA 3D visualization software (Thermo, Waltham, MA) and displayed as a Z-axis projection. Before imaging, spheroids were fixed with 4% paraformaldehyde for 10 min at room temperature, then permeabilized with 0.1% Triton X-100 for 15 min at room temperature (Sigma). Following permeabilization, spheroids were stained with Griffonia Simplicifolia-1 Isolectin 4 conjugated to FITC (GS-1) (Vector Laboratories, Burlingame, CA) at a dilution of 1:500. Concomitantly, mouse monoclonal α-smooth muscle actin primary antibody was added at 1:250 (α-SMA) (Santa Cruz Biotechnology, Dallas, TX). Spheroids were incubated overnight at 4°C. The following day, spheroids were washed three times with PBS and incubated with RedDot nuclear stain (Biotium, Fremont, CA) and goat anti-mouse IgG Alexa Fluor 594 secondary antibody (Thermo Fisher, Waltham, MA) at 1:200 and 1:1000 dilutions, respectively, for 2 h at room temperature. Samples were subsequently washed with PBS and imaged as aforementioned. Endothelial components stained green with GS-1 FITC. Perivascular support cells as well as fibroblastic components stained red with α-SMA conjugated to Alexa Fluor 594, and nuclei stained with RedDot were artificially colored blue.
Statistical analysis
One-way ANOVA with Dunnett's multiple comparison post-test was used to determine statistical significance between mean spheroid diameters at day 2 and all other subsequent time points through GraphPad Prism 7 for Windows (GraphPad Software, Inc., La Jolla, CA).
Results
The 3D bioprinter utilized in these studies was capable of time and pressure-regulated extrusion of materials at various viscosities using a range of extrusion pen tips. Measured contact angles for water and a suspension of cells, media, and collagen at printed concentrations were recorded at 155° and 156°, respectively. Table 1 illustrates the range of printing parameters tested. The parameters selected represent the conditions that provided consistent extrusion of spheroids with uniform size at a rate of 10.3 spheroids/min. Syringe pressure, pressure duration, and needle gauge, all combine to determine extrusion speed and, therefore, spheroid size. Higher pressures and larger needle gauges allow for more consistent results and were selected for those reasons. Pressure duration was kept low to maintain spheroid volumes. Cartridge size was selected to best fit our batch volumes and collagen concentration was selected to maintain the spheroid properties compared with unprinted collagen gels. We have previously published the conditions necessary to extrude cell-laden collagen cylinders with maintenance of cell viability and these reported values were used as a starting point for the current studies.41,42
Pressure effects evaluated using 100 ms pressure duration.
Time of pressure extrusion evaluated at a constant 5 psi syringe pressure.
The nScrypt Bioassembly Tool utilizes EFD cartridges in a precision fluid dispensing system (Nordson Corporation, Westlake, OH).
Nordson EFD extrusion tips.
Rat tail-derived collagen.
To maintain spheroid morphology after collagen extrusion, but before collagen polymerization, we utilized a superhydrophobic coating on tissue culture polystyrene plates and 3D bioprinted spheroids directly onto these surfaces. As illustrated in Figure 1a, b, the spheroids are immobilized on the superhydrophobic surface by Pluronic F-127 throughout stage movement (X–Y axes) during printing and subsequent transport to an incubator for gelation. This Pluronic F-127 disk on the superhydrophobic surface is illustrated in the diagram in Figure 1d. Spheroids could then be easily released from the Pluronic F-127 either by incubation in an aqueous solution or by cooling the surface to 10°C. Throughout the entire process, spheroids maintained their shape on these superhydrophobic surfaces as well as after removal.
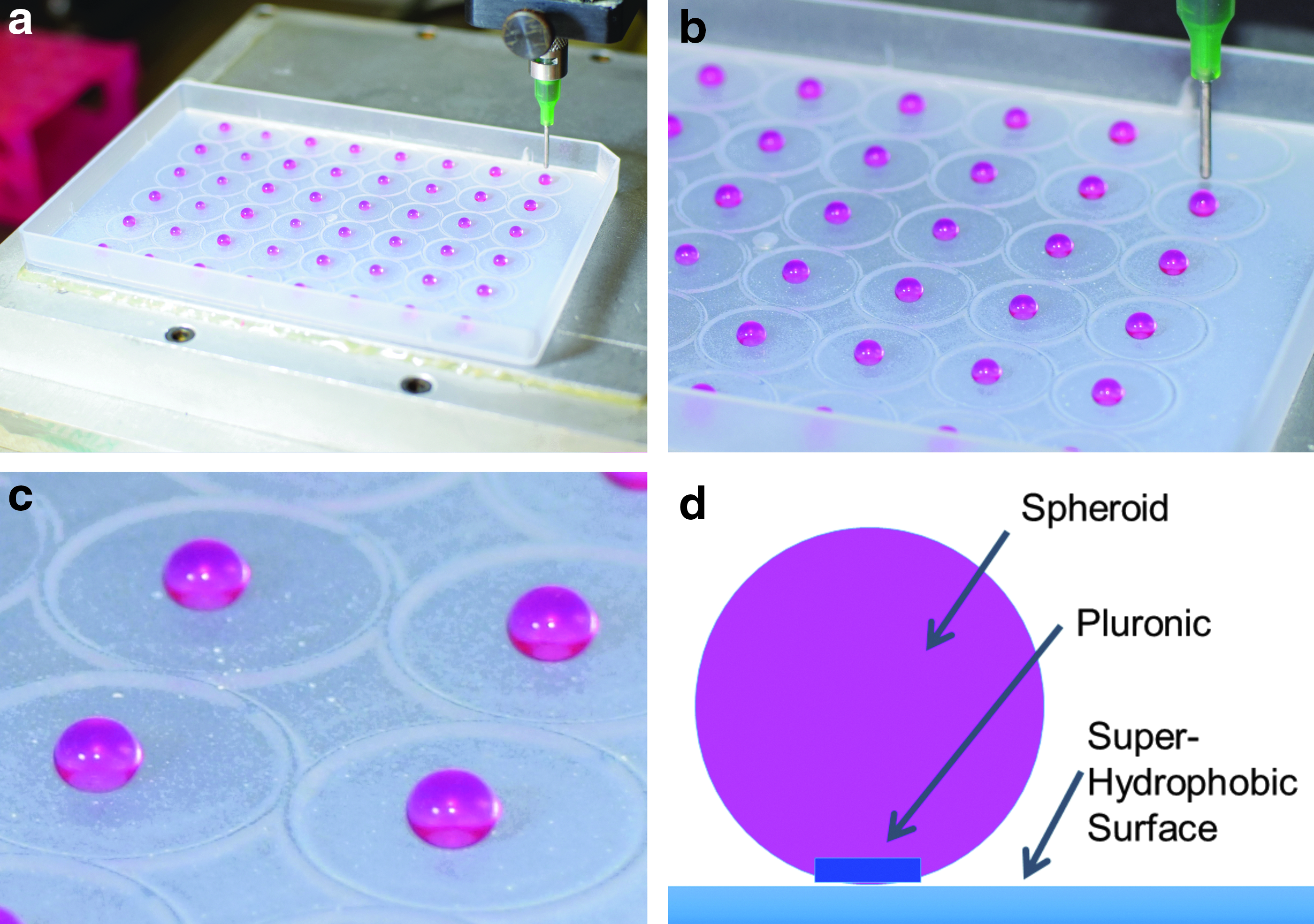
SVF cell-laden collagen I spheroids immediately after bioprinting on a biphilic surface
Batches of spheroids were prepared using 2 mL of unpolymerized collagen I containing suspended SVF cells, and were produced in quantities of 100–150 spheroids per batch depending upon size. Spheroids had a mean diameter of 3.54 mm with a standard deviation of 0.195 mm. Batch production time required 20 min of instrument preparation and 5 min per 48 spheroids extruded. After extrusion, the spheroids were incubated for 10 min at 37°C resulting in a total production time of 30–35 min before spheroid usage.
The typical morphology of 3D bioprinted SVF spheroids after initial collagen gelation is illustrated in Figure 2. The SVF cell-laden spheroid morphology, visualized by phase contrast microscopy, is uniform (Fig. 2a), and the engrafted cells are mostly viable as demonstrated by calcein AM live stain (Fig. 2b). A few nonviable cells, labeled with ethidium homodimer-1 were present as seen in Figure 2c. The distribution of cells assessed by nuclear staining with bis-benzimide (Fig. 2d) was homogeneous indicating that the gelation of collagen maintains cell encapsulation throughout the spheroid with no evidence of cell settling within a particular location. The distribution and viability of cells within the spheroids were assessed again after 6 days in spinner culture. Live cells (calcein AM positive) are seen in Figure 2f, with dead cells (ethidium homodimer-1 positive) shown in Figure 2g, and total cell distribution by nuclear staining (bis-benzimide positive) shown in Figure 2h. At this culture time point, morphological changes in the encapsulated SVF were noted, including microvessel structures with elongated sprouts and complex geometries suggesting the occurrence of in vitro angiogenesis in 3D collagen I.

Spheroid morphology visualized by phase contrast microscopy
SVF viability was quantified immediately after printing on day 0 and throughout the culture period on days 2, 6, 9, and 13 in Figure 3. Cell viability was quantified using standard curves provided in Supplementary Figures S1 and S2 (Supplementary Data are available online at

Cell viability within spheroids on days 0, 2, 6, 9, and 13.
Studies of the spheroids during suspension culture indicated that spheroids undergo contraction with concomitant reduction in diameter. The contraction of the spheroids is illustrated by phase contrast microscopy in Figure 4 and was quantitatively assessed with results provided in Figure 5. Early collagen contraction occurs at an average rate of 0.0083 ± 0.0875 mm/day from days 2 to 3 and the rate of contraction increases to its peak rate of 0.3675 ± 0.1359 mm/day from days 6 to 7 in spinner culture. Following this peak, contraction rate decreases to 0.042 ± 0.0463 mm/day from days 9 to 13 in spinner culture. Average spheroid diameters were significantly different between days 2 and all subsequent days in culture (Fig. 5, p < 0.0001).

Phase contrast micrographs of an SVF cell-laden collagen I spheroid on days 2

SVF cell-laden collagen spheroids show significant mean diameter decrease from 6 to 13 days of spinner culture incubation (n = 12). Statistical significance determined at p < 0.05. **p < 0.01, ****p < 0.0001.
The general morphology of the SVF spheroids was assessed at intermediate times up to 14 days in spinner culture and results illustrated in Figure 6. Due to the high density of cells in contracted spheroids highlighted in Figure 6B, neither phase contrast nor epifluorescence was adequate to produce clear images of cellular morphology in contracted spheroids. To visualize the distribution of cells and to characterize vascular cell phenotype in contracted spheroids, 14-day spheroids were stained for endothelial specific (GS-1 FITC positive) and anti α-smooth muscle actin antibody (α-SMA-Alexa Fluor 594 positive) and evaluated by confocal microscopy, which obtained images illustrated in Figure 6E–G. Amira software was used to create 3D volume renderings of the entire construct. The collective phase contrast and confocal images (Fig. 6A–G) indicate increased complexity of vessel-like structures as previously reported before in 3D collagen I in vitro assays utilizing SVF.21,44 An example of cellular components exhibiting vessel-like formation with the presence of endothelial cells luminally surrounded by α-SMA-positive cells, provides evidence of angiogenesis in vitro, and is highlighted by arrows in Figure 6F and G.
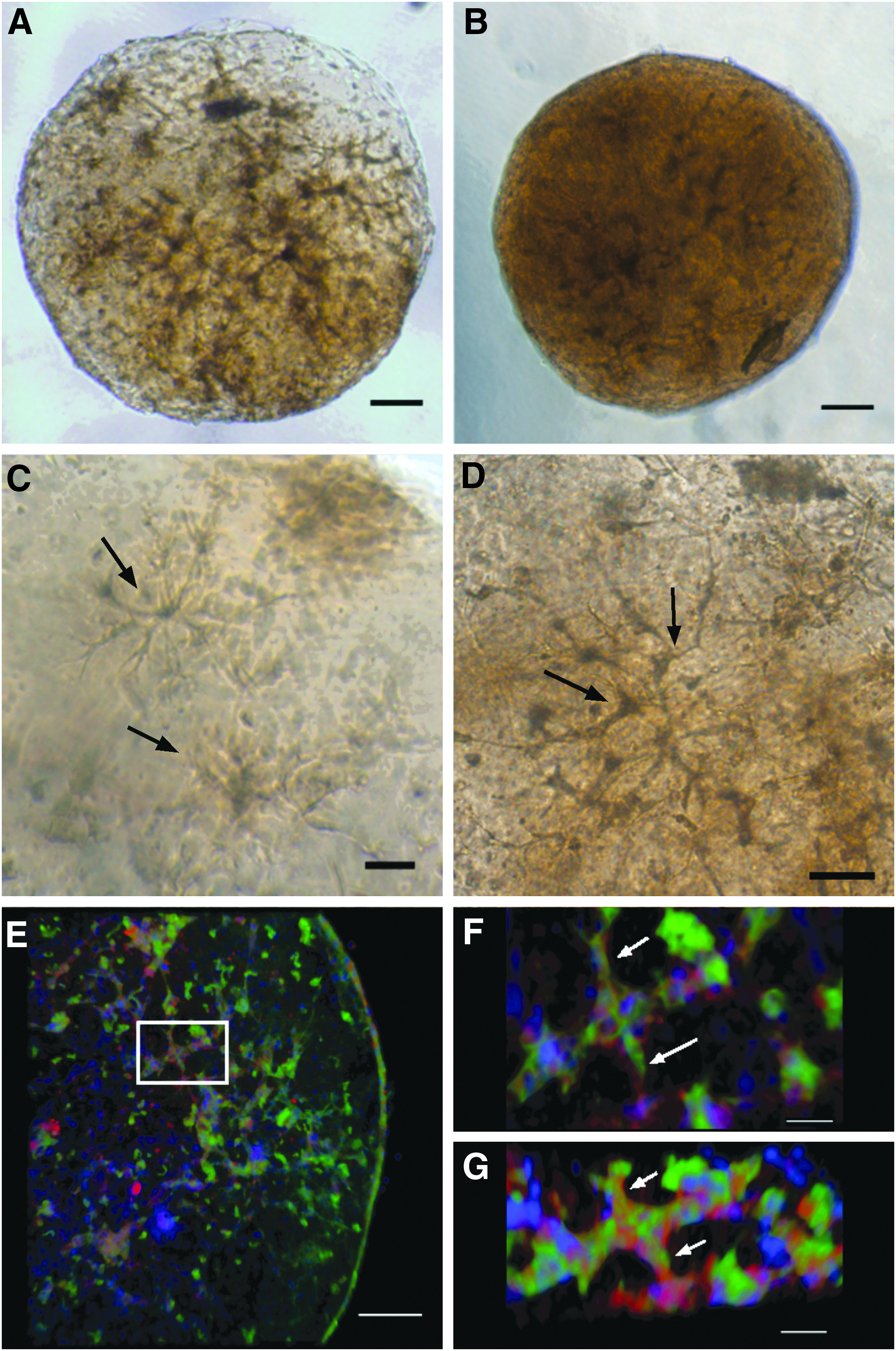
Phase contrast and confocal microscopy images of SVF spheroids.
Discussion
The delivery of stem and regenerative cells, including SVF, for the treatment of various diseases has reached human clinical trials. The limited retention of cells at the site of implantation has been implicated in observed poor therapeutic effect. 45 Alternative methods to improve retention have included the self-aggregation of cells in culture and the encapsulation of cells within a biomaterial.14,46 These studies have established an improvement in cell retention at the site of implantation. 45 SVF has been encapsulated in a variety of materials to improve cell retention, including alginate, to create SVF-containing spheroids.22,23 Retention of SVF in alginate maintains cell viability; however, alginate gels do not show any morphological change, which implies that they do not support engrafted cell migration and proliferation, thus limiting in vitro angiogenesis and potential in vivo application for cell-based therapies. For this reason, we explored the use of collagen type I for spheroid production as SVF cell populations have been shown to exhibit in vitro angiogenesis within this extracellular matrix (ECM).21,44,47 SVF cell-laden collagen gels can be created in sheets and are cumbersome to manipulate. Additionally, collagen spheroids printed onto conventional tissue plates rapidly flatten due to the hydrophilic characteristics of polystyrene. To maintain collagen I in a spheroid shape after extrusion and after polymerization, we utilized a superhydrophobic coating on tissue culture polystyrene plates and 3D bioprinted directly onto these surfaces.
Initial attempts to print onto superhydrophobic surfaces were unsuccessful at first due to the inability of spheroids to attach to the superhydrophobic surface, with most remaining attached to the printing pen tips. To overcome this lack of adherence, a disk of Pluronic F-127 was printed onto the superhydrophobic surface. Pluronic F-127 is a triblock copolymer with an amphiphilic block structure, which gives it hydrophilic and hydrophobic properties. This allows Pluronic F-127 to both adhere to the superhydrophobic surface and adhere to an aqueous material, such as collagen. With the biphilic surface established, collagen spheroids were produced using conditions that provided a variety of sizes. Mean diameter adjustments could be made through changes in applied pressure, which allowed the production of spheroids ranging in size from 1 mm to 3.5 mm.
In comparison to self-assembly based spheroid production methods, biphilic spheroid production is an order of magnitude faster with similar consistency in spheroid size and shape. 48 The primary source of spheroid size variation when using an automated time–pressure extrusion system, such as the BAT is the viscosity of the fluid being dispensed. However, all parameters, including stable neutral pH, low temperature, and thorough mixing of the unpolymerized collagen solution before BAT extrusion, were addressed to minimize variation within a spheroid batch.
These studies indicate that spheroid integrity and cell distribution are maintained throughout the 14-day cell culture period either in static culture or in more dynamic spinner culture conditions. The viability of the cells measured by the NucleoCounter before encapsulation in collagen was 96%, so the day 0 viability dropped by just under 30% during the process of collagen embedding. However, the viability within pipetted collagen was only 5.96% higher than that of the printed collagen, which leads to the conclusion that the collagen encapsulation rather than the bioprinting is the source of the viability decrease. This conclusion is in conflict with the prior research into the viability within pipetted collagen carried out by Smith et al. showing >90% viability for pipetted collagen and printed collagen. 42 Especially given that they used the same markers for their viability assays with cells of the same origin encapsulated in collagen of the same concentration. The two differences that most likely contribute to the viability difference seen are that Smith et al. cultured their cells before printing as opposed to our use of fresh isolate and our use of rat tail collagen, whereas their collagen source is not listed. This conflict could be attributed to the fact that Smith et al. did their cell counts for viability by eye rather than by fluorescence values. Counting cells for viability by eye leaves data vulnerable to user bias against dimmer objects. This issue is potentially exaggerated by the fact that in terms of relative fluorescence units, the live cells are an order of magnitude brighter than an equivalent number of dead cells and brightness compensation for image-based viability counts is frequently unregulated. This seems especially likely given that Smith et al. clarified that the cells had to be stained brightly in either color to be counted. This is further supported by the fact that the difference in viability between pipetted and printed cells is similar for both studies. A potential issue with fluorimeter-based viability is the possibility of nonspecific staining skewing results; however, our use of stained acellular collagen spheroids as baselines should have helped compensate for acellular signals and the standard curve should compensate for cellular nonspecific staining. Following day 0, viability remains high throughout the duration of the spinner culture, although there is a drop in viability that coincides with later stage spheroid contraction at day 9.
In contrast to results using alginate to form spheroids, 22 there is a noticeable change in cell morphology that demonstrates the ability of cells to undergo initial steps of angiogenesis, including but not limited to tip cell formation, sprouting and the formation of branch-like structures highlighted in Figure 6C and D. Contraction of the SVF collagen spheroids was also observed commencing after 6 days of cell culture and continuing throughout the culture period. Collagen contraction in the presence of fibroblasts and endothelial cells has been well documented and it has been suggested that cell-dependent contraction provides an assessment of active metabolism, microenvironment remodeling, and cellular motility of these embedded cells. 49 Confocal microscopic analysis of cellular composition and phenotype in contracted spheroids (Fig. 6E–G) suggests the presence of organized, elongated, vessel-like structures that stain positive for the endothelial cell-specific lectin Griffonia simplificiolia-1 (GS-1) in conjunction with α-smooth muscle actin. This phenotypic change over time provides evidence that SVF is either self-organizing into microvessel-like structures (vasculogenesis) or endothelial cells are sprouting from pre-existing microvessel fragments (angiogenesis). Angiogenesis and neovascularization of SVF in vitro provides therapeutic potential to utilize prevascularized collagen I spheroids for the treatment of ischemic conditions in vivo.
Automation through direct-write computer-assisted design and manufacturing provides the opportunity to produce spheroids in a controlled, precise, and efficient manner utilizing biphilic surfaces. Such systems also present the opportunity to design high-throughput microenvironment-based assays for a plethora of purposes, including drug delivery, cell delivery, or immunotherapy delivery. A number of in vitro applications of spheroids have yet to be investigated. The ability to produce a multitude of ECM-based spheroids composed of biomaterials, such as fibrin, hyaluronic acid, and other unexplored hydrogels would allow the creation of microenvironments suitable for a variety of cell-based therapies. Indeed, phenotypic variation and cellular function may be different depending upon the ECM used. 50 In vivo studies are also necessary to address and characterize the therapeutic benefit of encapsulated cells, including functional interaction with surrounding host tissue, in comparison to direct injection of liquid suspended cells alone.
In conclusion, this study demonstrates that a biphilic surface can be used to create viable SVF cell-laden spheroids of uniform size and shape. The addition of automation through a 3D bioprinter allows for high throughput and a production time low enough to fit within a point-of-care clinical setting. Furthermore, the use of a water-soluble hydrophilic spot and the phase transition properties of Pluronic F-127 allow for minimally disruptive spheroid removal for any applications that would require spheroid manipulation. These SVF cell-laden collagen spheroids may offer a strategy to increase the therapeutic effect of cellular infusions for regenerative purposes through improved cellular localization and retention.
Footnotes
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.