
Abstract
Extracellular matrix (ECM) hydrogels prepared by tissue decellularization have been reported as natural injectable materials suitable for neural tissue repair. In this study, we prepared ECM hydrogel derived from human umbilical cord (UC) and evaluated its composition and mechanical and biological properties in comparison with the previously described ECM hydrogels derived from porcine urinary bladder (UB), brain, and spinal cord. The ECM hydrogels did not differ from each other in the concentration of collagen, while the highest content of glycosaminoglycans as well as the shortest gelation time was found for UC-ECM. The elastic modulus was then found to be the highest for UB-ECM. In spite of a different origin, topography, and composition, all ECM hydrogels similarly promoted the migration of human mesenchymal stem cells (MSCs) and differentiation of neural stem cells, as well as axonal outgrowth in vitro. However, only UC-ECM significantly improved proliferation of tissue-specific UC-derived MSCs when compared with the other ECMs. Injection of UC-ECM hydrogels into a photothrombotic cortical ischemic lesion in rats proved its in vivo gelation and infiltration with host macrophages. In summary, this study proposes UC-ECM hydrogel as an easily accessible biomaterial of human origin, which has the potential for neural as well as other soft tissue reconstruction.
Introduction
T
In contrast to artificially designed tissue-engineered materials that fail to mimic the complex structure and chemistry of the cell microenvironment seen in vivo, biologic scaffolds comprising native extracellular matrix (ECM) represent structures very similar to those of the uninjured host tissue with many advantages, such as complex natural composition, three-dimensional structure, retention of growth factors, and bioactive properties, including stimulation of angiogenesis and migration of endogenous progenitor cells or modulation of immune reaction. 4 These unique features of ECM scaffolds ensure functional remodeling of the lesioned area, which is in contrast to scar tissue formation that usually takes place during standard healing. 5
Biological ECM scaffolds have been used for the reconstruction of numerous tissues, including myocardium, 6 skeletal muscles, 7 musculotendinous tissues,8,9 lung, 10 urinary tract,11,12 esophagus, 13 peripheral nerve, 14 or dura mater, 15 and have already been utilized for the treatment of many patients with tolerable tissue responses and positive clinical outcomes. 16
Both allogeneic and xenogeneic biological ECM materials are currently being transplanted for tissue and organ replacement. As xenografts, ECMs from nonprimate mammals (typically pigs) are commonly used, which might be associated with adverse host tissue inflammatory reactions due to the human immune response against mammalian ECM by anti-Gal and anti-nonGal antibodies. 17 Despite the fact that the role of α gal-specific antibodies in xenogeneic ECM implantation appears to be minimal,18,19 human-derived biomaterials could be more desirable as they avoid concerns related to potential immune responses as well as xenogeneic disease transfer.
Besides xenogeneic ECM, acellular allografts of human origin are being commonly prepared from cadavers and used, for example, as bone, valves, cornea, or skin grafts. 20 Remarkably, the age of the donor tissue is an important parameter that causes significant differences between ECM properties harvested from the same source. It has been shown that with age, there is an increasing ECM crosslinking due to nonenzymatic protein glycation, 21 adipose tissue deposition, and fibrosis, while biologic materials derived from younger animals are associated with a more constructive, site-appropriate tissue remodeling response than scaffolds derived from older animals.22,23 On the other hand, cadaveric tissues might differ in age of the human donors and are supposed to be considerably older than the porcine sources. For instance, comparison of human versus porcine tissue sources for an injectable myocardial matrix hydrogel revealed that despite the similarity of these matrices, there is an increased difficulty in the processing of human tissue and significant patient-to patient variability. 24
In contrast to adult tissue, the ECM from fetal or neonatal tissue comprises more immature collagen with few crosslinks, which promotes more effective tissue remodeling.25–27 It is therefore conceivable that ECM derived from fetal or neonatal tissue would induce more efficient and constructive tissue remodeling than ECM derived from adult or old tissues.
In this context, umbilical cord (UC) represents the suitable neonatal tissue source, which is easily accessible in sufficient amounts without any ethical constraints. Of note, UC is commonly used in regenerative medicine for isolation of mesenchymal stem cells (MSCs) and endothelial cells and it can also serve as a source for hyaluronic acid isolation. Being an extraembryonic tissue, it eliminates age-related impediments such as ECM remodeling, fibrosis, oxidative stress, and other negative changes. 26
As a scaffold for neural tissue repair, injectable in situ gelling hydrogels have been considered more appropriate than fibrous ECM as these materials can easily conform to lesion irregularity with minimal tissue damage during delivery. To form injectable hydrogels, ECM from various decellularized tissues can be enzymatically solubilized to a liquid form, which self-assembles into a hydrogel under physiological pH and temperature.27–29 Previously described injectable ECM hydrogels prepared by decellularization of porcine brain (B-ECM), spinal cord (SC-ECM), and porcine urinary bladder (UB-ECM) revealed in vitro neurotrophic properties.30,31 Moreover, ECM hydrogels from UB-ECM and SC-ECM proved to be beneficial for providing a supportive environment in vivo after injection into an experimentally induced cavity in stroke 32 or spinal cord injury. 33
In this study, we propose human UC as a suitable source of ECM and introduce an optimized decellularization protocol to prepare UC-derived ECM hydrogel (UC-ECM). UC-ECM hydrogel is characterized in terms of its structure, composition, mechanical properties, and neurotrophic potential in vitro and compared with previously described UB-ECM, SC-ECM, and B-ECM. In addition, UC-ECM hydrogel was injected into cortical photothrombotic lesions in rats to prove its in situ gelation and in vivo biocompatibility.
Materials and Methods
Tissue decellularization and preparation of ECM hydrogels
Porcine UBs, spinal cords, and brains were obtained from an abattoir (Český Brod, Czech Republic); the age of the animals was 6 months. ECM hydrogels were prepared according to previously described protocols.30,34 Human UCs were obtained from healthy full-term neonates after spontaneous delivery with the informed consent of donors, using the guidelines approved by the Institutional Committee at University Hospital (Pilsen, Czech Republic). About 10–15 cm of umbilical tissues were frozen (>16 h at −20°C), aseptically transported into the laboratory, and subsequently thawed and transversely cut into pieces (<0.5 cm length). Tissue pieces were agitated in 0.1 M phosphate-buffered saline (PBS; IKEM, Czech Republic) bath (48 h at 120 rpm, 4°C). The PBS bath was exchanged three to five times before the tissue pieces were soaked in 0.02% trypsin/0.05% EDTA (120 min at 120 rpm, 37°C) and afterward in 0.1% peracetic acid in 4.0% ethanol bath (120 min at 300 rpm; Penta, Czech Republic), and in a series of PBS and deionized water (dH2O) soaks. Finally, tissue pieces were lyophilized for 24 h (FreeZone® 2.5; Labconco Corporation) and powdered (Mini-Mill Cutting Mills; Thomas Scientific). For the in vivo application, the powdered ECM was sterilized in ethylene oxide at 37°C overnight.
To prepare the hydrogel, powdered ECM samples were solubilized with 1.0 mg/mL pepsin in 0.01 N HCl (Sigma) at a concentration of 10 mg ECM/mL and stirred at room temperature for 48 h to form a pregel solution (pH ∼2). The pepsin-HCl ECM solution was neutralized to pH 7.4 with 0.1 N NaOH, isotonically balanced with 10 × PBS, and diluted with 1× PBS to the final concentration of 8 mg/mL, which allows in vivo gelation.32,33 To form the hydrogel, the neutralized pregel was placed at 37°C for ∼45 min.
ECM hydrogel characterization
Nanoscale topography
The surface topography of UC-ECM, UB-ECM, SC-ECM, and B-ECM hydrogels was analyzed by scanning electron microscopy (SEM) mode, applying previously published methodology. 35 Briefly, a gel was placed on the glass slide, fixed in cold 2.5% glutaraldehyde (Electron Microscopy Sciences) for 24 h, and washed in PBS. Hydrogels were then dehydrated in a graded series of alcohol, followed by subsequent chemical drying with hexamethyldisilazane (Sigma). The dried samples were cut to expose their inner structure and used for SEM studies. Micrographs were taken using an FEI Quanta 3D FEG scanning electron microscope at an acceleration voltage of 2 kV to prevent charging of unconductive samples. ImageJ (Rasband, W.S.; U.S. National Institutes of Health, Bethesda, MD) software was used for image processing and micrograph quantification. Using special plug-in for ImageJ (FibrilTool), 36 image analysis was performed in terms of anisotropy score calculation.
Efficiency of tissue decellularization
The absence of cell nuclei in decellularized tissues was proved by hematoxylin and eosin (H&E) staining. The nuclei were stained using 4′,6-diamidino-2-phenylindole fluorescent dye (DAPI, 1:1000; Invitrogen). Double-strain DNA (dsDNA) was isolated from native and decellularized tissue according to the manufacturer's instructions (DNeasy® Blood & Tissue Kit; Qiagen) and quantified using a spectrophotometer (NanoPhotometer™ P-Class). DNA content was normalized to the initial dry weight of the samples. For each native and decellularized tissue, the assay was repeated three times. Base pair length of residual DNA was determined on 2% agarose gel (Sigma) containing 0.5% SYBR Safe DNA Gel Stain (Thermo Fisher Scientific) and visualized with ultraviolet transillumination using a reference 50-base pair (bp) ladder (Cleaver Scientific).
Immunohistochemical analysis
To visualize the content of acid mucopolysaccharides, collagen, laminin, and fibronectin, the decellularized ECM was fixed in 4% paraformaldehyde in PBS and embedded in paraffin blocks. Sections of 5 μm thickness were mounted onto slides, deparaffinized, and stained with Alcian blue (pH 2.5; all acid mucosubstances, blue or greenish blue) together with Weigert's iron hematoxylin (nuclei, black) and Van Gieson's staining (acid fuchsin in saturated aqueous picric acid; collagen—red). In addition, immunohistochemistry using primary antibodies against collagen I (COL-I) (mouse monoclonal IgG1, clone COL-I, 1:1000), laminin (rabbit polyclonal IgG, 1:200), and fibronectin (rabbit polyclonal IgG, 1:200, all from Abcam) was performed. Goat anti-mouse IgG conjugated with Alexa Fluor 488 (1:400) for collagen, goat anti-rabbit IgG conjugated with Alexa Fluor 488 (1:400) for fibronectin, and goat anti-rabbit IgG conjugated with Alexa Fluor 594 (1:400) for laminin (all from Life Technologies) served as secondary antibodies. Images were taken using the LEICA CTR 6500 microscope (Leica Microsystems).
Collagen and glycosaminoglycan quantification
The collagen content in ECM hydrogels was assessed using colorimetric assay Sircol™ Insoluble Collagen Assay Kit (Biocolor Ltd.). Sulfated glycosaminoglycan (sGAG) concentrations in ECM hydrogels were determined using the Blyscan Sulfated Glycosaminoglycan Assay Kit (Biocolor). Absorbance of the samples was recorded at 555 nm for collagen and at 656 nm for sGAG content using a Tecan Spectra plate reader (Tecan). All assays were performed according to the manufacturer's recommended protocol from three independent samples measured in triplicates. The collagen and sGAG content was normalized to the initial dry weight of the samples.
Rheometry
Dynamic oscillatory shear tests were used to investigate the viscoelastic properties of ECM hydrogels. ECM hydrogels were subjected to a sinusoidal deformation in a 40-mm parallel plate rheometer (AR-G2; TA Instruments) at 1 Pa stress and 10°C to determine their mechanical response (displacement or strain) as a function of time. A dynamic time sweep was run with the parameters of 5% strain, 1 rad/s (0.159 Hz), and increasing the temperature from 10°C to 37°C to induce gelation as indicated by a sharp increase and plateau phase of the storage modulus (G′). The assay was repeated three times with three independent samples in triplicates.
Turbidity gelation measurement
The turbidimetric gelation kinetics were determined on a spectrophotometer (Infinite® 200 Pro; Tecan), which was preheated to 37°C. ECM hydrogel samples (8 mg/mL) were kept on ice at 4°C until 100 μL was pipetted into each well of a 96-well plate and inserted into a spectrophotometer. Absorbance was measured at 405 nm every 10 min for 100 min. Normalized absorbance time to reach 50% and 95% maximal absorbance were determined as t1/2 and t95. 37 The lag time (tlag) was defined as the point where a line representing the slope at log t1/2 intersects the turbidimetry baseline with 0% absorbance. The gelation rate (S) was defined as the slope of the linear region of the gelation curve. The measurements were repeated three times with three independent samples in triplicates.
In vitro characterization on cell culture
Mesenchymal stem cells
Human bone marrow (hBM) MSCs were obtained from Bioinova, Ltd. (Czech Republic), and cultured as described previously 38 in complete medium comprising MEM Alpha (Lonza) media, 5% platelet lysate (Bioinova, Ltd.), and gentamicin (10 μg/mL; gentamicin Lek®; Lek Pharmaceuticals).
Human adipose tissue-derived MSCs (hASCs) were obtained from healthy volunteers who underwent liposuction procedures for esthetic reasons and signed an informed consent. The lipoaspirate was repeatedly washed in PBS and enzymatically digested by collagenase (0.3 PzU/mL, Collagenase NB 6 GMP; Serva Electrophoresis GmbH) at 37°C, centrifuged at 1000 rpm/min for 5 min, and the cells were cultured under standard conditions.
Human Wharton's jelly (hWJ) MSCs were collected from fresh human UCs as described in the Tissue Decellularization and Preparation of ECM Hydrogels section. About 10–15 cm of UC was aseptically transported into sterile PBS with antibiotic–antimycotic solution (Sigma) at 4°C. After removal of blood vessels, the remaining tissue was chopped into small pieces (1–2 mm3) and transferred to 10-cm Nunc culture dishes (Schoeller, Czech Republic) containing the complete medium. On day 10, the explants were removed from the culture dishes and remaining adherent cells were cultured for 3 weeks or until 90% confluence.
All hMSCs were cultivated at 37°C and 5% CO2 in a humidified atmosphere; the medium was changed twice a week. Cells in the third passage were analyzed for surface markers and used for the ECM hydrogel evaluation.
Using a flow cytometer (FACSAria™; Becton Dickinson), all three types of MSCs were proved, for characteristic surface markers, positive for CD90, CD73, CD29, CD10, CD44 (Exbio, Czech Republic), CD105 (BioLegend), and HLA ABC (BD Pharmingen) and negative for CD14, CD34, CD45 (Exbio), CD133 (Miltenyi Biotec), VEGFR2 (BioLegend), CD31, and HLA-DR (Pharmingen). Data analysis was performed using BD FASCDiva software.
Human neural stem cells
A conditionally immortalized human fetal neural stem cell (NSC) line SPC-01 was generated from 8-week-old human fetal spinal cord as described previously. 35 Cells were cultured in tissue culture flasks, freshly coated with laminin (10 μg/mL) in DMEM/F12 (Gibco, Life Technologies), and supplemented with human serum albumin (0.03%; Baxter Healthcare Ltd.), human apo-transferrin (100 μg/mL), putrescine DiHCl (16.2 μg/mL), human recombinant insulin (5 μg/mL), progesterone (60 ng/mL), L-glutamine (2 mM), sodium selenite (40 ng/mL), 4-OHT (100 nM) (all from Sigma), human EGF (20 ng/mL), human bFGF (10 ng/mL) (PeproTech), and primocin 100 μg/mL (InvivoGen) at 37°C and 5% CO2 in a humidified atmosphere.
Cell growth and proliferation
To determine in vitro MSC proliferation, ECM hydrogels were placed into a 96-well plate (90 μL/well) and seeded with cells (5000 cells/cm2 in 100 μL media). Cells seeded on wells without hydrogel (tissue culture plastic) served as a control. Cell proliferation was measured using WST-1 assay (Roche) after 1, 3, 7, and 14 days of the culture. Ten microliters of WST-1 reagent was added to each well containing 100 μL culture media, and the plates were incubated for 2 h at 37°C. The absorbance was measured using a Tecan Spectra plate reader at 450 nm. Each type of hydrogel was seeded in triplicate. Six independent experiments of three hydrogel batches were performed for each hydrogel type.
The morphology of MSCs grown on hydrogels was examined by immunofluorescence staining for actin filaments. After fixation in 4% paraformaldehyde in PBS for 15 min, the cells were washed with PBS and stained with Alexa Fluor 568 phalloidin (1:400; Molecular Probes); the nuclei were visualized using DAPI fluorescent dye (1:1000; Invitrogen).
NSC growth and differentiation were analyzed after their seeding (100,000/cm2) on laminin-coated coverslips or ECM hydrogel disks formed inside a cylindrical mold with a diameter of 0.8 cm (Scaffdex). After 7 and 14 days of culture, the cells were fixed in 4% paraformaldehyde in PBS for 15 min, washed with PBS, and stained using Alexa Fluor 568 phalloidin. Next, immunohistochemical staining against light neurofilaments (70 kDa) using mouse monoclonal IgG1 (NF70, 1:400; clone DA2, Merck Millipore) and mouse monoclonal IgG against microtubule-associated protein 2 (MAP2, 1:1000; clone AP20; Millipore) was performed. The nuclei were visualized using DAPI fluorescent dye (1:1000, Invitrogen). Images were taken using the confocal microscope Zeiss LSM 5 DUO (Carl Zeiss, MicroImaging GmbH).
Cell migration
To assess the chemotactic capacity of ECM hydrogels for hMSCs, xCELLigence® RTCA DP Instrument was used to perform Cell Invasion and Migration Assay (Acea Biosciences, Inc.). ECM hydrogels (6% v/v solutions in media without supplements) were placed into a lower chamber of CIM-Plate 16, and hBM-MSCs, hASCs, and hWJ-MSCs (105 cells in 100 μL of media without supplements) were then pipetted into an upper chamber. The impedance signal was reduced when cells adhered to the microporous membrane to migrate toward ECM hydrogels. As a positive control, chemokine stromal cell-derived factor 1 (SDF-1, 20 ng/mL; Sigma) was used; culture medium without supplements served as a negative control. Recorded impedance signal was then used to assess the rate of cell migration. The assay was repeated five times.
Dorsal root ganglia explant culture
Dorsal root ganglia (DRGs) were extracted from four 3–5-day-old Wistar rats (Velaz, Czech Republic). Briefly, the spinal cords were dissected and DRGs from low thoracic and lumbar parts were isolated, placed in cold Hank's balanced salt solution without Ca2+/Mg2+ solution (Invitrogen), and cleaned of peripheral nerve processes. DRG explants were then placed on ECM hydrogels or Matrigel (Matrigel® Growth Factor Reduced Basement Membrane Matrix, Phenol Red-Free; Corning) in 24-well plates, and cultured in neurobasal medium (Invitrogen) supplemented with 2% B27 (Life Technologies), 2 mM L-glutamine (Invitrogen), 0.5% NGF (50 ng/mL; PeproTech), uridine (17.5 μg/mL; Sigma), and primocin (2 μL/mL; PeproTech) in a humidified atmosphere at 37°C and 5% CO2. The medium was changed every 3 days. After 7 days of culture, DRGs were fixed with 4% paraformaldehyde in PBS for 10 min and stained for anti-NF160 antibody (clone NN18, 1:200; Sigma), Alexa Fluor 488 secondary antibody (1:200; Invitrogen), and cell nuclei (DAPI, 1:1000; Invitrogen). Fluorescent images were taken using the Leica fluorescence microscope (Leica DMI 6000B) and TissueGnostic software (TissueGnostics GmbH). The neurite extension area and the longest neurite length were determined using a special plug-in for ImageJ (NeuriteJ). 39 The assay was repeated three times for each hydrogel.
Injection of UC-ECM hydrogels into a rat model of cortical photothrombotic lesion
To test biocompatibility of UC-ECM hydrogel in vivo, a focal brain photochemical lesion was created in the motor cortex in rats. Eight male Wistar rats (330 ± 30 g) were maintained at 22°C on a 12-h light/12-h dark schedule and given water and food ad libitum. To cause the lesion, the animal was placed into the stereotactic apparatus under isoflurane (3%) anesthesia, the scalp incision was created in the midline, and the pericranial tissue was dissected to expose the bregma. Focal cerebral ischemia was performed according to 40 as follows: Bengal Rose (Sigma) was injected through the right femoral vein (0.08 g/mL saline; 1 μL/g of the animal weight), and the skull was illuminated above the primary motor cortex (2 mm rostral and 2 mm dextrolateral to the bregma) with a fiber-optic bundle of a cold light source (KL 1500 LCD; Zeiss) for 10 min. The skin overlying the cranium was then sutured.
Seven days after the focal cerebral ischemia, a small opening in the lesion site was drilled into the scull of the animal and 10 μL of UC-ECM (n = 4) or saline (n = 4) was injected into the lesion (in a depth of 2 mm) using a Hamilton syringe (Hamilton Company, Bonaduz, Switzerland) and stereotactic apparatus.
Animals were sacrificed 24 h after the implantation with an overdose of anesthesia and perfused with 4% paraformaldehyde in 0.1 M PBS intracardially. The brains were removed, fixed in 4% paraformaldehyde for 10 days, and cut in frozen mode (local temperature −24°C). Coronal slides, 40 μm thick, were stained for cell nuclei with DAPI (1:1000; Life Technologies), mouse monoclonal IgG1 to CD68 (ED1, 1:150; Abcam), goat polyclonal IgG to CD206 (c-20; 1:250; Santa Cruz), or mouse monoclonal IgG1 to COL-I (1:1000, COL-I; Abcam) diluted in 0.1 M PBS containing goat (or donkey–depending on the host organism of secondary antibodies) serum (1:10 both; Sigma) and Triton X-100 (0.1%) overnight in 4°C. Staining solution lacking Triton X-100 was used only in the case of extracellular anticollagen staining. As secondary antibodies, goat anti-mouse IgG conjugated with Alexa Fluor 594 (1:400) for COL-I, donkey anti-mouse IgG conjugated with Alexa Fluor 488 (1:400) for CD68, and donkey anti-goat IgG conjugated with Alexa Fluor 594 (1:400) for CD206 (all from Life Technologies) were used.
Fluorescent images were taken using confocal microscope Zeiss LSM 5 DUO. The relative number of macrophages in the hydrogel area was determined from three randomly selected sections using a 20 × objective and ImageJ software.
All experiments on animals were performed in accordance with the European Communities Council Directive of 24th November 1986 (86/609/EEC) regarding the use of animals in research and were approved by the Ethics Committee of the Institute of Experimental Medicine Academy of Sciences, Czech Republic, Prague.
Statistical analysis
Data are presented as mean ± standard error of mean. The statistical significance was analyzed using one-way ANOVA with Tukey's multiple comparison post hoc analysis (GraphPad Prism) with a level of p < 0.05 considered statistically significant.
Results
Structure of ECM hydrogels
B-ECM, SC-ECM, and UB-ECM hydrogels were prepared by decellularization protocols as described previously.30,34 Due to a high amount of hyaluronic acid, which causes massive tissue swelling in water, the decellularization procedure of UC-ECM required more washing steps in dH2O and PBS to thoroughly remove the cells from the ECM. Importantly, we found that agitation of the tissue in trypsin/EDTA was a necessary step to achieve gelation of the resultant ECM hydrogel (Fig. 1A, B).
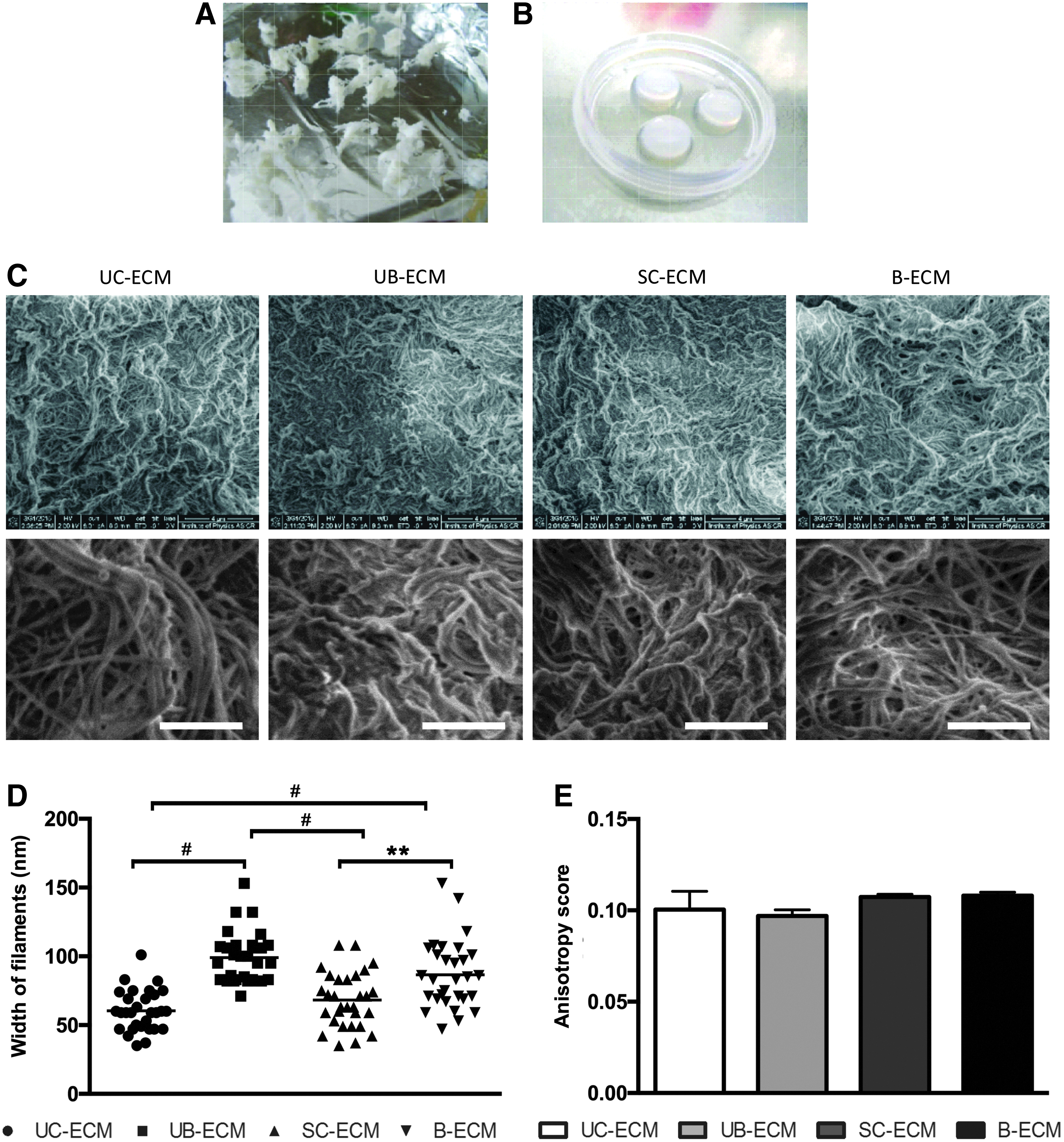
The fiber network topology of different ECM hydrogels was examined on SEM images (Fig. 1C). Microscopic comparison of these gels shows that fiber width of UC-ECM (60.47 ± 2.73 nm) and SC-ECM (68.23 ± 43.61 nm) was significantly smaller in comparison with UB-ECM (99.00 ± 3.37 nm) and B-ECM (86.57 ± 4.53 nm, Fig. 1D). To extract quantitative data on fibril orientation, circular statistics were used, which are adapted to directional data, to analyze the properties of the tangent direction over the region of interest. The circular variance of the tangent direction defines the score, determining whether the fibrils are well ordered (fibril array anisotropy, Fig. 1E). With regard to the anisotropy score, the following conventions were used: 0 for no order (purely isotropic arrays) and 1 for perfectly ordered, that is, parallel fibrils (purely anisotropic arrays). 36 Indeed, there was no difference with regard to the anisotropy score between all types of ECMs (Fig. 1E).
Composition of ECM hydrogels
Similarly to porcine ECMs, UC-ECM was successfully decellularized with minimal cellular content within the scaffold (Fig. 2). H&E and DAPI staining confirmed the absence of residual cell nuclei (Fig. 2A). Quantification of dsDNA showed that in all ECM samples, the residual dsDNA was less than 50 ng per mg dry ECM (Fig. 2B, C). Previous studies4,32,41 show that DNA contained in ECM should not exceed 50 ng/mg of the tissue in order not to elicit an immune reaction on the recipient's site. Our protocol of UC decellularization ensured efficient cell removal, and resulting UC-ECM contained <50 ng or no residual dsDNA, and therefore met the basic requirement for clinical application.
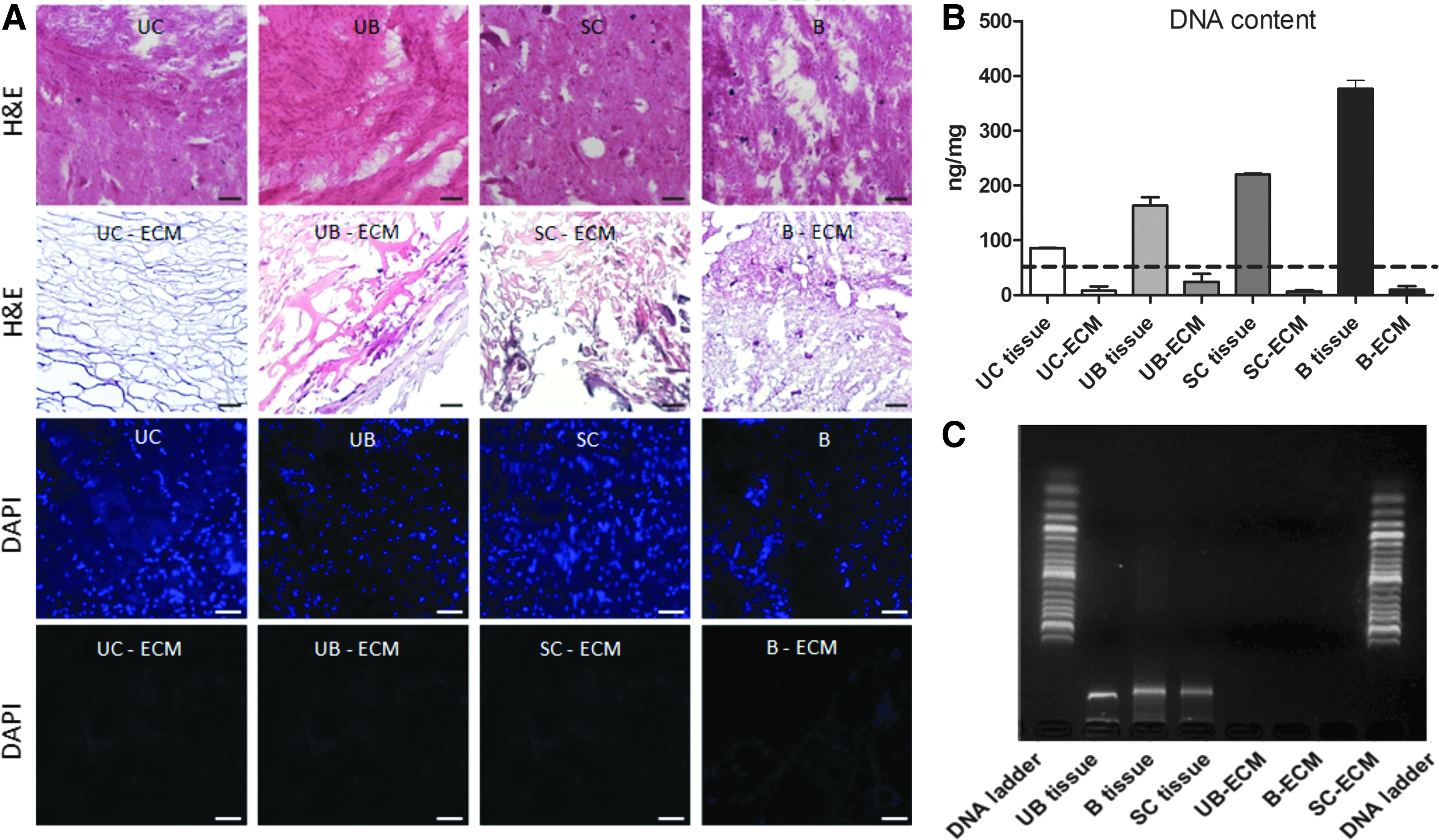
An important parameter, which shows efficacy of ECM harvesting, is the ECM yield, expressed as the percentage of dry weight of ECM to wet weight of the initial source tissue (Table 1). The highest ECM yield was found for UB-ECM 8.06% ± 4.66% (n = 6), followed by UC-ECM 0.96% ± 0.51% (n = 6). In contrast, the decellularization of CNS tissues, which requires more complex decellularization procedures due to the high amount of myelin, resulted in a very low ECM yield; 0.25% ± 0.14% for SC-ECM (n = 6) and 0.12% ± 0.07% for B-ECM (n = 6), respectively.
ECM yield is expressed as a percentage of the dry weight of ECM to the wet weight of the initial tissue.
ECM, extracellular matrix.
Alcian blue was used to stain acid polysaccharides such as GAGs (Fig. 3A). Immunohistochemical staining showed that ECM scaffolds comprise collagen (Fig. 3B), laminin (Fig. 3C), and fibronectin (Fig. 3D).
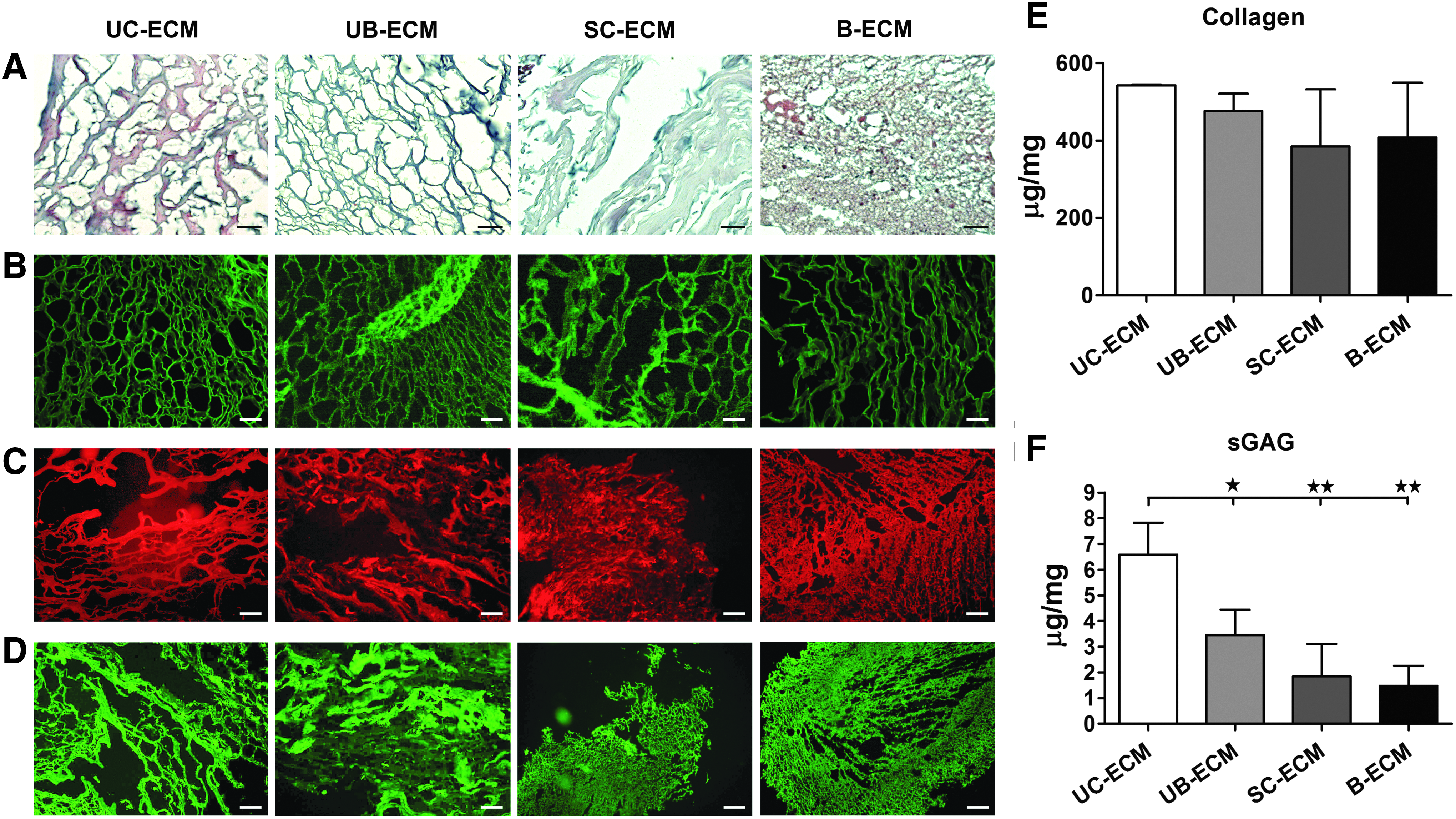
Collagen concentration did not significantly differ among all the ECM types. The highest amount of collagen was found for UC-ECM (542.6 ± 1.78 μg/mg of dry ECM weight), then for UB-ECM (476.1 ± 45.38 μg/mg), B-ECM (408.1 ± 141.0 μg/mg), and SC-ECM (384.7 ± 147.6 μg/mg) (Fig. 3E).
It has been shown that the concentration of hyaluronic acid within ECM is high in fetal and newborn tissues, including UCs. 42 Indeed, the significantly highest amount of sGAG was found in UC-ECM (6.59 ± 1.24 μg/mg) when compared with UB-ECM (3.46 ± 0.99 μg/mg), SC-ECM (1.84 ± 1.26 μg/mg), and B-ECM (1.47 ± 0.79 μg/mg of dry ECM weight) (Fig. 3F).
Rheological and turbidimetric measurements
In rheological experiments, we determined the flow and mechanical properties of ECM hydrogels. Increasing the strain, G′ of ECM hydrogels, started a decline of 10% amplitude when they became more viscose, which was observed for all ECM hydrogels, irrespective of their origin (Fig. 4A). The storage modulus of the matrix was found to be the highest for UB-ECM, while the softest material was B-ECM.

Turbidimetric gelation kinetic curves showed a sigmoidal shape for UC-ECM, whereas other ECMs had an exponential shape (Fig. 4B). A significantly longer lag phase as well as the longer time required to reach half of the final turbidity (t1/2) and 95% of the final turbidity (t95) was found for UB-ECM than for other ECM hydrogels, while the shortest gelation time was found for UC-ECM (Table 1 and Fig. 4B–E). However, the velocity to complete gelation or gelation rate (S) was significantly higher for SC-ECM when compared with UB-ECM (Fig. 4F). These results suggest that hydrogel assembly was fastest for UC-ECM, followed by SC-ECM and B-ECM, while the UB-ECM hydrogels had the longest gelation time.
Cell growth, proliferation, and migration on ECM hydrogels
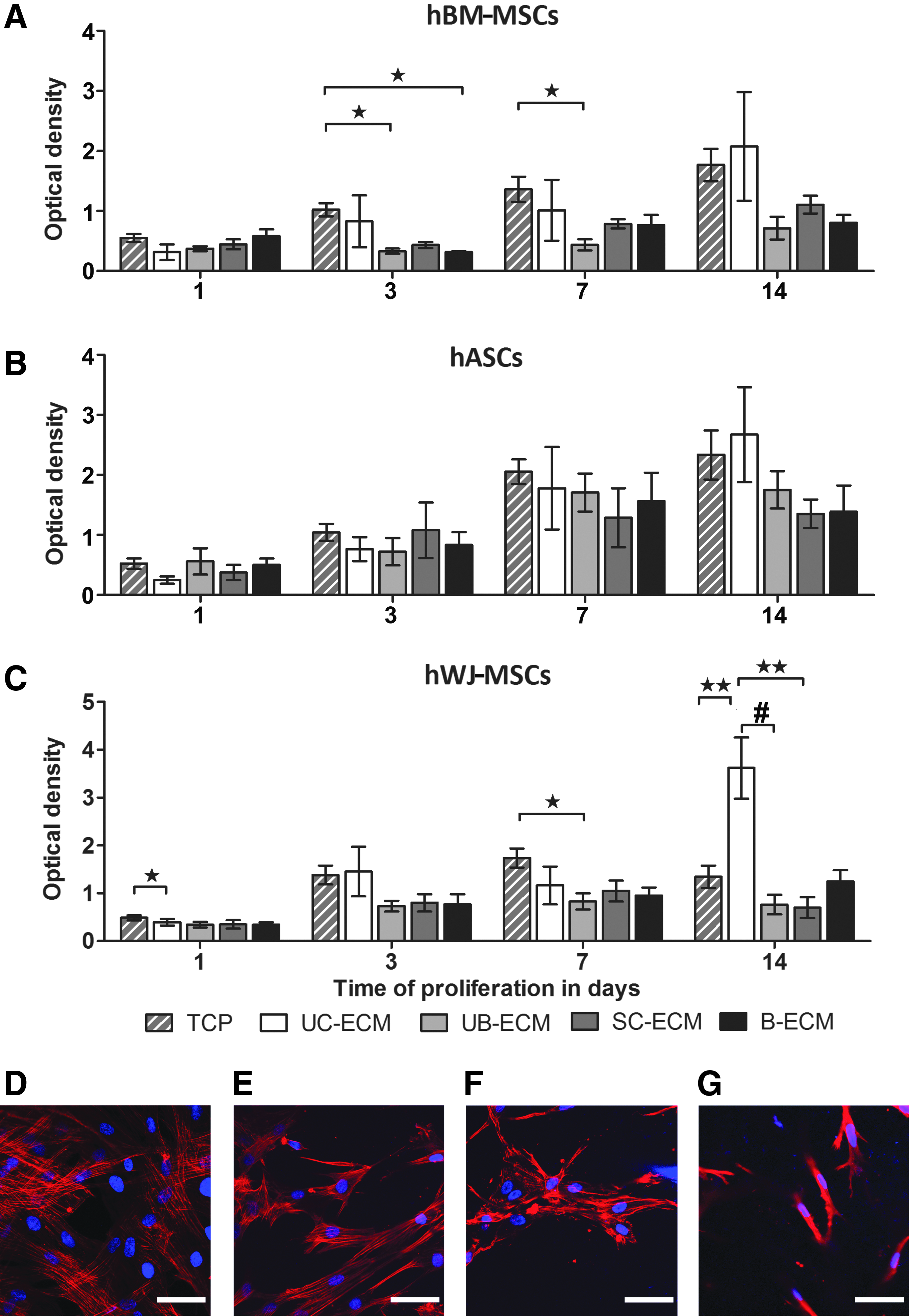
Proliferation of hBM-MSCs, hASCs, and hWJ-MSCs on ECM hydrogels was determined using WST-1 assay after 1, 3, 7, and 14 days of the culture. While BM-MSCs on UC-ECM proliferated similarly as in the control tissue culture well, lower proliferation of BM-MSCs was found on all porcine-derived hydrogels, UB-ECM, SC-ECM, and B-ECM. On the other hand, proliferation of hASCs on all types of ECM hydrogels did not significantly differ from controls. Notably, proliferation of hWJ-MSCs at 14 days was significantly higher on tissue-specific UC-ECM than on other ECM hydrogels or even in controls (Fig. 5).
Using migration assay, all ECM hydrogels revealed chemotactic properties and stimulated the migration of all types of MSCs, which was significantly higher when compared with cell migration to the control culture medium alone (Fig. 6A–C).

Chemotactic properties of ECM hydrogels investigated using xCELLigence® RTCA DP Cell Invasion and Migration Assay showing migration of
The ability of ECM hydrogels to support neurite outgrowth was observed using DRG explant cultures. After 7 days of culture, neurites densely extended from DRG bodies with no significant differences in neurite length or neurite area between individual hydrogels and Matrigel, which served as a positive control (Fig. 6D–F).
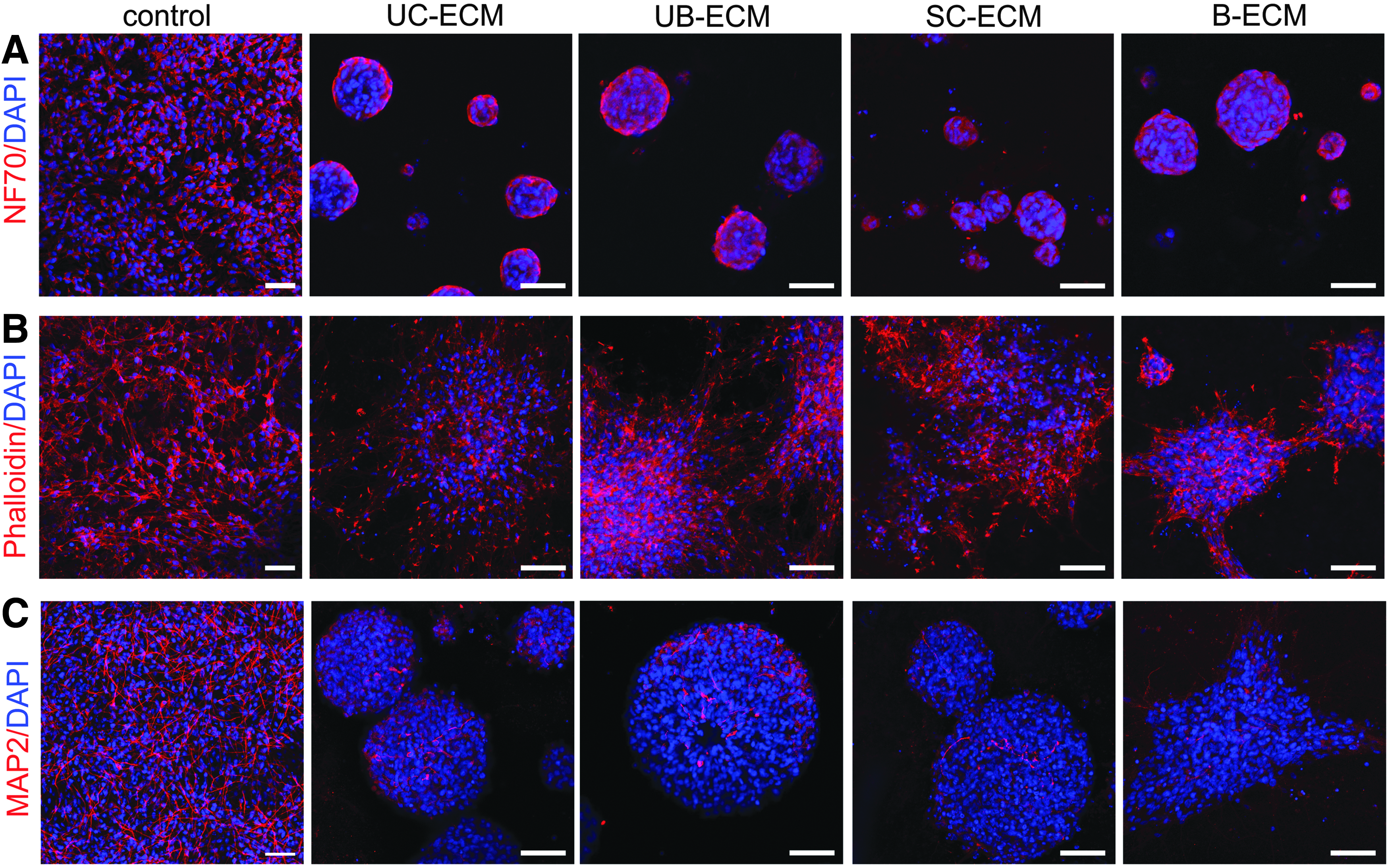
It is well known that the unique compositions and microstructural features of ECM have been shown to be influential in directing cell fate or morphology. 43 To reveal the neurotrophic properties of ECM hydrogels, we determined neural differentiation of NSC culture. NSCs grew on all ECM hydrogels, and after 7 days, displayed an expression of the early neuronal marker NF70 (Fig. 7). Of note, in contrast to the dispersed NSCs cultured on laminin-coated glass coverslips, the NSCs grown on the ECM hydrogels tend to grow in distinct clusters with delimitated borders. After 14 days in culture, NSCs proliferated and differentiated into neuronal cells positive for neuronal marker MAP2, without any remarkable differences between particular ECM hydrogels (Fig. 7).
In vivo evaluation of ECM hydrogels
To prove the in vivo gelation and biocompatibility of UC-ECM, the hydrogel was injected into the focal ischemic lesion created in the rat motor cortex. As is illustrated in Figure 8 on collagen and DAPI staining after 24 h, the UC-ECM formed a compact hydrogel within the lesion, which was highly populated by endogenous cells. The host macrophages were the prevalent cell type present within the lesion and also infiltrating the hydrogel (75.7% ± 5.0% of all infiltrating cells in the gel, n = 3). As is apparent from Figure 8C, the macrophages detected within the UC-ECM hydrogel revealed predominant positivity for marker of M2 macrophages (CD206+ cells), which represented 77.1% ± 6.5% of all macrophages within the hydrogel.

Coronal brain sections illustrating in vivo gelation and cellular infiltration of UC-ECM hydrogel 24 h after implantation into the photothrombotic ischemic lesion in the rat motor cortex.
Discussion
Acellular tissue-specific ECMs can be utilized as biological scaffolds for many applications in tissue engineering. For reconstruction of neural or other soft tissues, ECMs in the form of hydrogels are clinically more acceptable as these materials retain biologic activity, with the advantage of injectability and in situ polymerization, which offer minimally invasive delivery techniques. Indeed, injectable ECM hydrogels derived from different tissue sources have been previously demonstrated for the treatment of spinal cord injury, 33 stroke, 27 postmyocardial infarction, 44 critical limb ischemia, 45 and new adipose tissue development. 46
In this study, we optimized the decellularization protocol as described by Medberry et al. 30 to prepare an injectable hydrogel from human UC tissue, which combines the advantages of neonatal tissue of human origin with the ease of availability, without any ethical or regulatory constraints. In contrast to other previously used ECMs extracted from the porcine UB, brain, or spinal cord, UC-ECM contained a significantly higher amount of sGAG, which is generally high in fetal and newborn tissues, including the UC. 42 Moreover, the sGAG present in ECM includes chondroitin sulfates, heparin, heparan sulfate, and hyaluronic acid, and these structures can bind cytokines and growth factors such as bFGF and contribute to neural tissue reconstruction.34,47–50
The concentration of sGAG has been shown to alter gelation kinetics and mechanical properties of ECM hydrogels. 30 Indeed, UC-ECM revealed the fastest gelation rate when compared with porcine ECMs with lower sGAG. Notably, even if the B-ECM, SC-ECM, and UB-ECM were prepared by the same decellularization protocols, they displayed slight differences in collagen and sGAG concentrations as well as in gelation time and rheologic properties when compared with those described by Medberry et al. 30 These differences might result from different origin as well as the age of porcine tissues used for decellularization.
It has been suggested that ECM scaffolds derived from the same tissue type as that of the injury site may have a unique composition of molecular constituents to induce constructive tissue-specific remodeling. For instance, cardiac ECM has demonstrated the capability to provide tissue-specific cues for cardiac cell growth and differentiation. 6 Tissue-specific skeletal muscle ECM revealed better outcomes for critical limb ischemia treatment compared with UC-ECM. 45 In addition, the B-ECM hydrogel, being the tissue-specific matrix, unlike the SC-ECM and UB-ECM, increased the length of neurite extensions of the neuroblastoma cell line. 30
On the contrary, other studies have not shown an advantage of CNS-derived ECM versus non-CNS-derived ECM materials with respect to their effects on in vitro neurite outgrowth34,41 or in vivo neurotrophic properties in spinal cord injury repair. 33
Similarly, in this study, despite dissimilarities in nanoscale topography, sGAG content, mechanical properties, and the speed of gelation, we did not observe any significant differences in chemotactic or neurotrophic properties between the CNS- and non-CNS-derived ECM materials of human or porcine origin, which does not indicate an advantage of tissue specificity for ECM hydrogels in neural tissue repair. Of note, to achieve effective cell removal, CNS-derived tissues required the more complex decellularization procedures compared with, for example, UB-ECM or UC-ECM, which may result in the removal of neurosupportive proteins and growth factors and consequent loss of CNS tissue-specific bioactivity.
On the other hand, we found a tissue-specific effect of UC-ECM, which selectively promoted proliferation of hWJ-MSCs when compared with the other types of ECMs and MSCs.
A significant, but neglected, feature of ECM hydrogels is their contraction in 3D fibroblast-like culture. This well-known phenomenon is a characteristic of collagen gels seeded with fibroblasts, which generate tension on the matrix during both extension and retraction of pseudopodia.
51
A similar effect has also been described for ECM hydrogels derived from porcine dermis and UB seeded with fibroblasts.
52
We previously reported that porcine SC-ECM and UB-ECM significantly contracted when combined with hWJ-MSCs in a 3D culture
33
; in this study, we demonstrated contraction also for UC-ECM and B-ECM. The contraction rate, however, occurred slower for UC-ECM and B-ECM in comparison with SC-ECM and UB-ECM (Supplementary Data; Supplementary Data are available online at
When used as an MSC vehicle in vivo to fill the lesion cavity, rapid gel contraction may then result in an inhomogeneous scaffold distribution within the lesion, which should be taken into consideration, when applying ECM hydrogels as cell carriers in in vivo settings. On the other hand, other cell types such as NSCs can be cultured in ECM hydrogel without causing contraction.
Finally, we proved in vivo UC-ECM gelation and biocompatibility using a rat model of photothrombotic lesion. The UC-ECM was densely infiltrated by resident macrophages with a predominating M2-like phenotype. An M2-like macrophage phenotype was also found using SC-ECM and UB-ECM hydrogel in rat spinal cord injury 33 and UB-ECM injected into rat middle cerebral ischemia lesion cavities. 32 An M2-positive macrophage infiltration in response to acellular ECM has been positively correlated with constructive host tissue remodeling, while M1 phenotype resulted in the deposition of dense connective tissue and scarring.25,53 Nevertheless, future work is needed to determine the in vivo UC-ECM degradation and its replacement by the host tissue in a longer time period.
In summary, our results expand the previously described ECM hydrogels and have proved UC-ECM to be an easily accessible material of human origin that provides appropriate mechanical and bioactive properties suitable for CNS repair.
Conclusion
In this study, we prepared injectable ECM hydrogel by decellularization of human UC tissue and compared its properties with CNS and non-CNS ECM hydrogels derived from porcine UB, brain, and spinal cord. Despite differences in nanoscale topography, sGAG content, and speed of gelation, all ECMs revealed similar in vitro neurotrophic properties, which do not indicate a benefit of tissue-specific ECM for neural tissue repair. After injection into the cortical photothrombotic lesion in rats, the UC-ECM formed a hydrogel in situ and was densely populated by host macrophages. Further in vivo studies are needed to consider the potential of ECM hydrogels for clinical translation.
Footnotes
Acknowledgments
This work was supported by GACR 15-01396S, GAUK 1846214, Ministry of Education, Youth and Sports of CR within the LO1309, BIOCEV (CZ.1.05/1.1.00/02.0109), CZ.02.1.01/0.0./0.0/15_003/0000419, and the J.E. Purkyně fellowship awarded by Academy of Sciences of the Czech Republic. The authors thank Stephen F. Badylak and Christopher J. Medberry for the initial training and providing the decellularization protocols.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.