
Abstract
For many years, the impact of hyper- and hypothermia on mammalian cells has been examined. With the exception of short, low temperature storage, which has uses in areas such as preservation for transplantation or regenerative medicine, advantages for the use of low temperature treatment in hepatocytes have not been previously reported. We have observed that alginate-encapsulated HepG2 liver spheroids that are cryopreserved or experience a cold reduction in temperature (≤10°C) for periods between 1 and 90 min display an enhanced cell proliferation during culture 7–16 days post-treatment compared with untreated samples. Following 8–12 days post-treatment, alginate-encapsulated liver spheroids experienced a cell density of 1.71 ± 0.35 times that of control samples (p < 0.001). This effect occurred in samples with a variety of cold treatments. This low temperature treatment offers a simple method to rapidly increase cell proliferation rates for extended culture systems, such as bioartificial liver devices. This would allow the manufacture of required biomass more rapidly, and to a higher cell density, reducing final required biomass volume. This could enable bioartificial liver devices to be prepared more cheaply, making them a more cost effective treatment.
Introduction
A
The cryopreservation of these ELS has been studied previously,8,9 and groups have examined the cryopreservation of other alginate-encapsulated cell lines.7,10,11 This has been done either to preserve ELS once a sufficient cell density has been achieved, or to explore the possibility that alginate encapsulation may offer some cryoprotection. It was in the course of our work that we observed a stimulatory effect on cell proliferation within ELS following freezing and thawing.9,12–14 In this study, the potential for low temperatures to induce proliferation of alginate-encapsulated cells upon return to culture conditions has been examined in detail. We studied both the impact of chilling (low temperature exposure without freezing) and freezing (ultralow temperature in the presence of ice) in the presence of protective agents such as dimethyl sulfoxide (Me2SO or DMSO) or those found in Viaspan.15–20
Materials and Methods
Cell culture and encapsulation
Cell culture—static ELS culture
ELS were added to T175 flasks filled with warmed culture medium of modified alpha-minimum essential medium, supplemented with 50 U/mL penicillin, 50 μg/mL streptomycin (all Invitrogen, Paisley, United Kingdom), 0.5% 1 M CaCl2 (v/v; Sigma, Gillingham, United Kingdom), and 10% human blood plasma (National Blood Transfusion Service) at an ELS medium volumetric ratio of 1:32. The ELS were cultured in a 37°C, 5% CO2 humidified incubator. One hundred percent medium changes were carried out every 2–3 days.
Encapsulation
HepG2 cells were cultured in monolayer (ECACC, Wiltshire, United Kingdom). Each experiment used a fresh aliquot from our Working Cell Bank at our GMP cryobanking facility (Fisher Bioservices). These cells were expanded and passaged twice. At reaching 80–90% confluence after the second passage, cells were detached and passaged a third time for encapsulation. An aqueous solution containing 2% (w/v) alginate (Manugel; FMC BioPolymers, Philadelphia, PA) and 1.5% (w/v) 10–50 μm glass beads acting as a buoyancy regulator (Kisker BioTech, Steinfurt, Germany) was mixed volumetrically 1:1 in culture medium containing 4 million cells/mL. This gave an approximate final solution of 1% alginate, 0.75% glass beads, and 2 million cells/mL. 6
This mix was passed through an encapsulation system (Genialab Jetcutter, Braunschweig, Germany), producing spherical droplets with a targeted 500 μm radius that was cross-linked in 0.204 M CaCl2 solution. This resulted in spheroids with individual cells distributed internally. The actual achieved alginate bead size and cell density on encapsulation is shown below in Table 1, which varied due to factors such as alginate viscosity, solution surface tension, and cells being lost in washing steps. These individual cells develop into spheroids, although the number of ultimate spheroids is lower due to some initial cell death and separate spheroids merging. Figure 1 shows microscopy of spheroids inside beads as cells proliferated over 12 days.
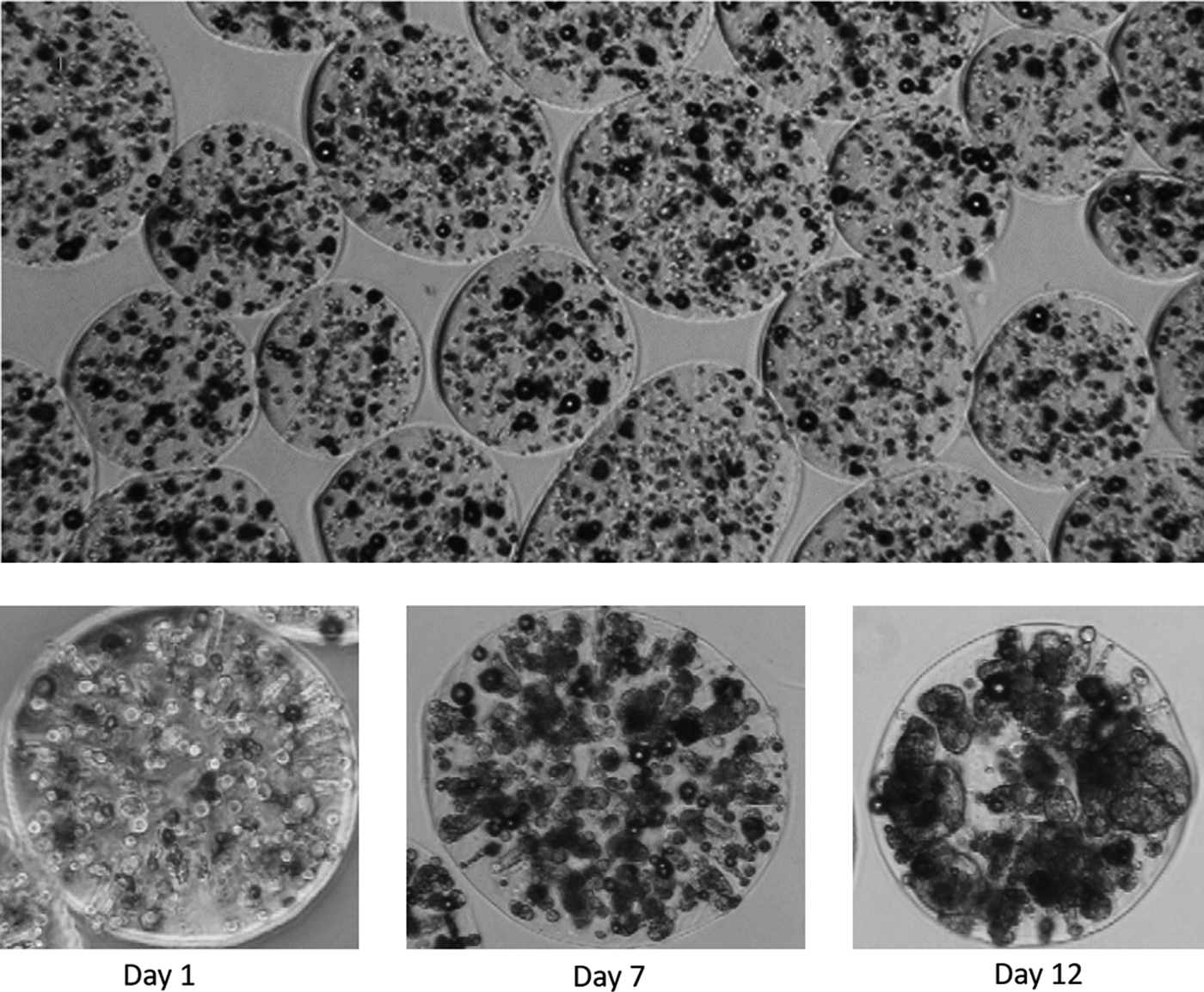
Top, an image of ELS immediately following encapsulation, showing the size and shape range of alginate-encapsulated HepG2 cells. The lower images show a single alginate bead at 1 day postencapsulation (1.92 million cells/mL), 7 days postencapsulation (8.9 million cells/mL), and 12 days postencapsulation (17.9 million cells/mL). The proliferation of single cells into spheroids is apparent. ELS, encapsulated liver spheroids.
This, combined with the measured average bead size has been used to determine the initial number of cells per alginate bead at the start of the culture period.
The encapsulation method has been described previously. 6 The entire process is carried out at room temperature.
Cooling/warming methods
Chilling
Unless otherwise stated, low temperatures were induced by plunging the samples (1 mL solutions of ELS with 1 mL excess medium in 2 mL cryovials) into the center of an ice bucket. Care was taken to ensure the whole sample was covered in ice and samples were not agitated during the chilling test. Upon completion of the test time (1–90 min), samples were removed from the ice bucket and transferred into 37°C culture medium before reculture.
For cooling rate studies, samples were cooled as above, or in an EF600 freezer (Asymptote Ltd., Cambridge, United Kingdom) at 0.3°C min−1 from 20°C to 0°C. Rapid cooling was achieved by pipetting 1 mL samples of ELS directly into 9 mL culture medium precooled at 0°C, to induce classical cold-shock.15,21,22 To test a 10°C chilling condition, samples were placed into an EF600 sample holder that had been precooled and maintained at 10°C.
To determine temperatures in cryovials, a dummy solution containing 10% glycerol (G5516; Sigma) in 0.15 M NaCl was prepared and added to cryovials with an inserted thermocouple (Picotechnology, St. Neots, United Kingdom). This was attached to a Picologger (Picotechnology) and recorded using a computer. This solution has previously been determined to have the same thermal properties as ELS in culture medium. 9
Reagent studies
In experimental conditions where the impact of reagents on cooling was explored, reagent solutions were prepared at 2 × concentration in 1 mL of culture medium, before addition to 1 mL sample resulting in a 2 mL final volume. Samples immediately underwent testing after mixing.
Cryopreservation protocol—Me2SO with Viaspan
Low temperature treatment induced by cryopreservation was examined through cooling ELS to 4°C, and mixed volumetrically 1:1 at 4°C with a solution containing 24% Me2SO (D4540; Sigma) in a 76% Viaspan solution (v/v; Bridge-to-Life, Columbia, SC), containing 0.2% IceStart (Asymptote Ltd.) as a nucleating agent, giving a final concentration of 50% ELS, 12% Me2SO, 38% Viapsan, and 0.1% Icestart.
This solution was allowed to equilibrate for 5 min, before being filled into five separate cryovials, to a 2 mL final volume. The cryovials were then cooled from 4°C to −100°C at 0.3°C min−1 in an EF600 controlled rate freezer.
Upon completion of the cooling run, cryovials were transferred to liquid nitrogen storage.
Cryopreservation thawing protocol
Samples were removed from liquid nitrogen storage and thawed in a 37°C water bath until the last ice crystal had melted. This process took 330 s. The freezing mix was washed out in a stepwise manner using culture medium chilled to 4°C. 9 Warm culture medium (37°C) was added, and the ELS placed in culture at 37°C, in a 5% CO2 humidified incubator.
Post-thaw functional assessments
Viability
At designated time points, ELS were removed from culture and stained with 20 μL propidium iodine (PI) solution (1 mg/mL; Sigma) and 10 μL fluorescein diacetate (FDA) solution (1 mg/mL; Sigma) to view under a fluorescent microscope.
PI (red) only stains the nucleus of cells with a nonfunctional membrane, indicating dead cells, while FDA (green) only stains metabolically active cells. By comparing the intensities of PI and FDA emission using a calibrated macro on a phase-contrast microscope; the cell viability can be quantified by a method that has previously been outlined.9,12
Cell counts
Total cell number was determined using an NC-200 automated counting system (Chemometric, Allerod, Denmark). The ELS were liberated from alginate using a 16 mM EDTA solution (Applichem, Darmstadt, Germany) before being washed in phosphate-buffered saline (Sigma) and disaggregated by vigorous syringing through a 21G needle.
All cells were lysed in solution and the nucleolus stained with PI. This solution was drawn into the automated counter and the stained nuclei counted. As HepG2 cells are mono-nuclear, this was converted to a cell density for the total ELS present.
Metabolic activity assay
A tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay known as MTT (Invitrogen) was used to determine cell viability. About 0.5 mL of a 0.75% (w/v) MTT solution was added to a small volume of ELS in vials with lids porous to gas. The vials were transferred to a 37°C, 5% CO2 humidified incubator for 3 h to allow crystal formation. After incubation, 0.5 mL acidified (4 mM HCl) iso-propanol (W292907; Sigma) solution was added to stop the reaction and dissolve the crystals. This solution was added to a 96-well plate, 100 μL per well, and the absorbance at 570 nm of each well was determined with an ANTHOS III plate reader (Biochrom, Cambourn, United Kingdom). The absorbance values were normalized against a control sample.
Glucose consumption
Healthy HepG2 cells consume glucose during normal cell function. Samples of the culture medium were taken throughout the culture process, and the remaining glucose concentration measured with a GM7 reader (Analox, Stourbridge, United Kingdom) using oxidase enzyme reactions. This was then related to glucose consumption per sample.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assays (ELISAs) to determine alpha-1-fetoprotein (AFP) production in ELS was carried out on culture medium samples removed during the culture process. A sandwich ELISA using mouse monoclonal antibodies ab10071 and ab10072 (Abcam, Cambridge, United Kingdom) as a capture, and as a horseradish peroxidase-linked antibody respectively, with Applichem fetoprotein (A6935) used for a standard curve. The values were normalized either to cell density or ELS volume.
Imaging
A phase contrast microscope was used to visually inspect ELS, with images recorded using Nikon Imaging Software.
Statistics
Unless otherwise stated, figures are presented as an average of five replicates, ±1 standard deviation. p-Values were determined through an appropriate t-test, with significance determined at p < 0.001 unless otherwise stated. p-Values for raw data are stated in figure legends.
Results
Cold treatment in culture medium
Figure 2 shows a summary of 6 different experiments, each with 5 replicates per condition (except in Fig. 2, set C where 10 were carried out). For ELS experiencing chilling or cryopreservation on the day of encapsulation, viable cell number after 8 days (set C), or 12 days (Fig. 2, sets A, B, D, E, F) of culture is on average 171% ± 35% of controls p < 0.001. Comparing doubling time for the culture process, the cell number density in control samples doubled every 3.3 ± 0.8 days on average, while the chilled or cryopreserved samples cell numbers doubled every 2.5 ± 0.5 days on average. Inter-experimental variation in cell density occurs due to natural variation in the system, explaining the variations seen in control values, which has also been noted previously. 6 An increase in cell number was apparent following both cryopreservation and chilling for 45 min.
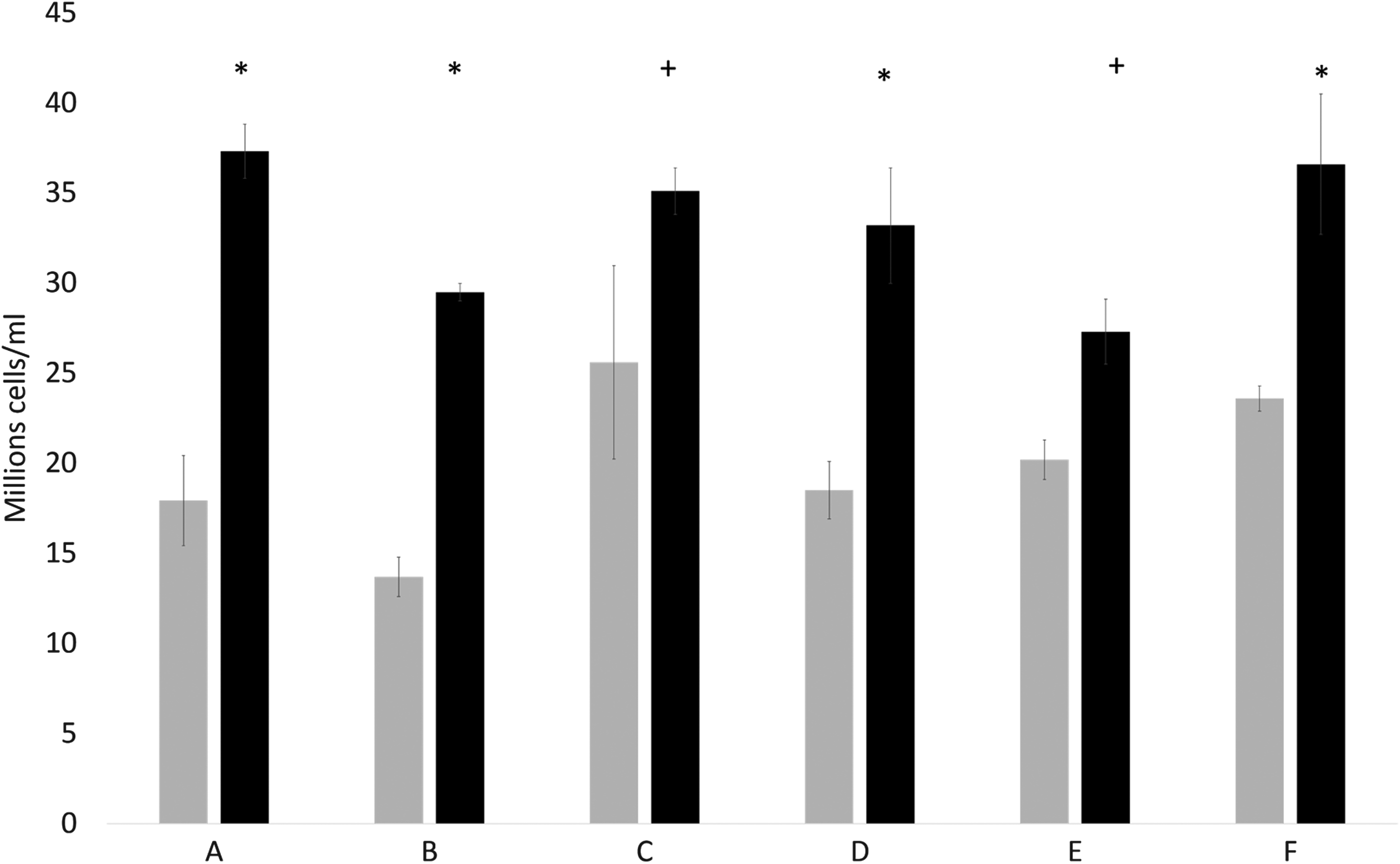
Comparison in viable cell number between samples following chilling or cryopreservation (black), and untreated control samples (gray). Data are presented as average of 5 samples ±1 SD, except set C where the control is average of n = 10. Sets A, B, and F experienced cryopreservation immediately post encapsulation, while sets C, D, and E were chilled to 0°C for 45 min immediately following encapsulation. All data are 12 days after treatment, except for set C that is 8 days. Initial cell concentration can be found in Table 1. All sets experience a significant improvement in performance, *indicated p < 0.001, +denotes p < 0.005. Combination of all sets A–F is significant at p < 0.001 using a two-tailed unpaired student's t-test. SD, standard deviation.
Impact of duration and temperature of cold exposure
Figure 3 demonstrates that increasing exposure of cells to low temperatures from 30 to 90 min does not significantly change the outcome between the sets. All sets achieved significantly improved cell numbers over the unchilled control (p < 0.001). Cell numbers after 8 days of culture were 19.1 ± 1.6, 30.7 ± 4.0, 34.6 ± 3.2, and 38.3 ± 3.5 million cells/mL for ELS samples experiencing no low temperatures, a 30-min hold at 0°C, a 45-min hold at 0°C, and a 90-min hold at 0°C, respectively.
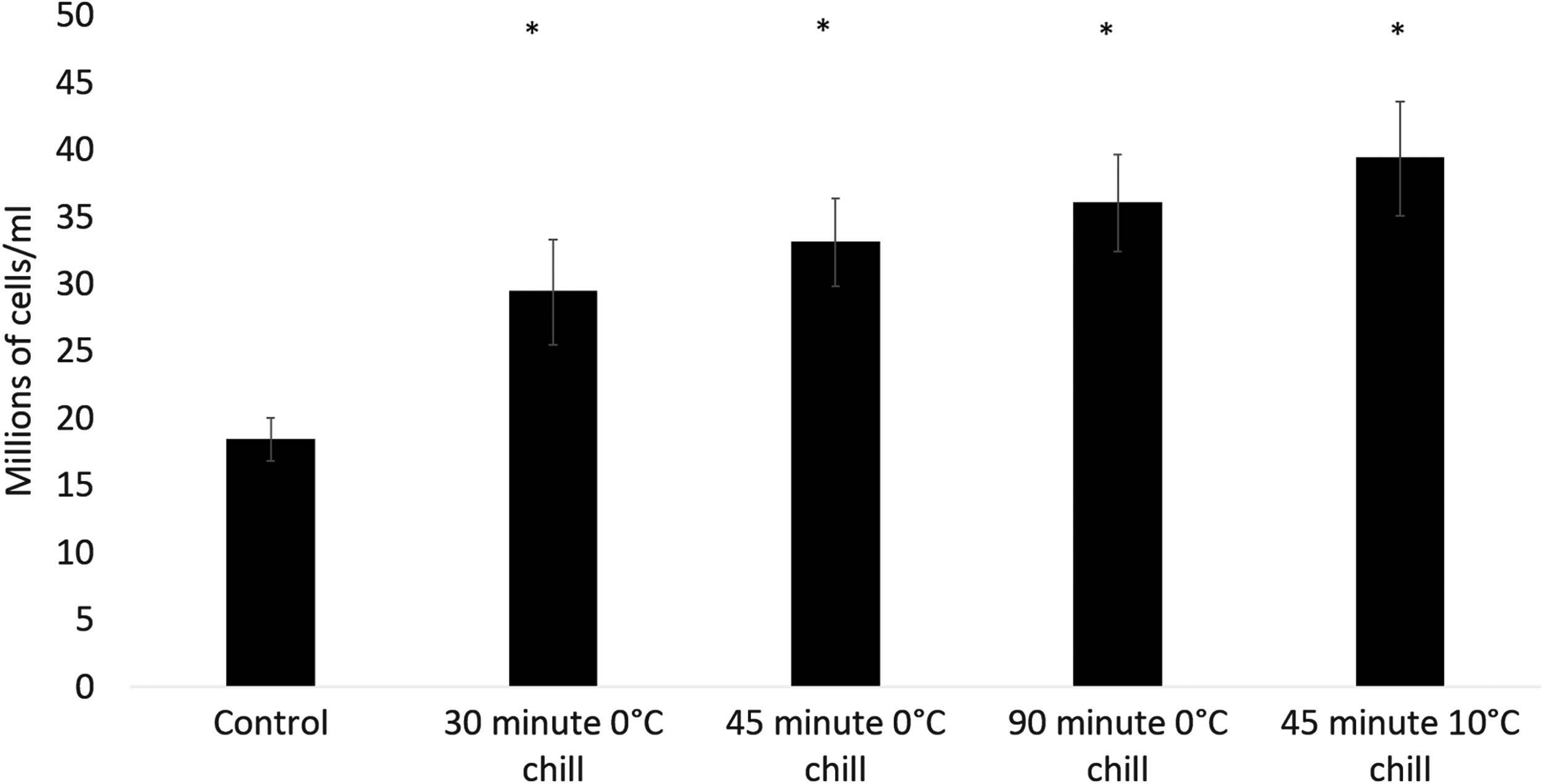
Viable cell number of samples experiencing various chilling times and temperatures, 12 days after treatment. Samples within 2 mL cryovials were either plunged into an ice bucket (0°C samples), or held at 10°C in an Asymptote EF600 controlled rate freezer immediately following encapsulation. All conditions (*) showed significantly higher viable cell number over the control at p < 0.001, using a two-tailed unpaired student's t-test, though no significant difference between sets was observed at that threshold. The initial cell concentration was 1.77 million cells/mL. Data are displayed as average of 5 ± SD.
When placed into an ice bucket, the sample temperature fell below 10°C after ∼80 s, and below 4°C after 3 min.
Cell viability was recorded as 96.5% ± 0.7%, 96.1% ± 0.5%, 95.9% ± 1.1%, and 94.3% ± 1.4% for ELS samples experiencing no low temperatures, 30-min hold at 0°C, 45 min at 0°C, and 90 min in an ice bucket, resulting in a viable cell density of 18.5 ± 1.6, 29.5 ± 3.8, 33.2 ± 3.2, and 36.1 ± 3.5 million cells/mL, respectively.
While the encapsulation process takes place at room temperature, Figure 3 also demonstrates that cooling samples to only 10°C for 45 min is sufficient to induce rapid cell growth, with a day 8 cell density of 19.1 ± 1.6 and 41.6 ± 4.3 million cells/mL for the control and cooled sample. This translates to a viable cell number 2.13 ± 0.22 times that of the control (p < 0.001).
Cooling rate and time dependency of low temperature treatment
To determine whether the rate of initial cooling was critical for the increased proliferation to become apparent, five conditions were tested:
(i) Plunging 2 mL samples within cryovials into an ice bucket, and holding for 30 min before rewarming. (ii) Plunging 2 mL samples within cryovials into an ice bucket, and holding for 60 min before rewarming. (iii) Pipetting 1 mL ELS into 9 mL precooled culture medium (at 0°C) to induce immediate cooling, and rewarming after 1 min. (iv) Pipetting 1 mL ELS into precooled culture medium (at 0°C) to induce immediate cooling, and rewarming after 30 min. (v) Cooling samples linearly from 20°C to 0°C at 0.3°C min−1 in cryovials in an EF600 controlled rate freezer.
Cell viabilities for each of the experimental conditions tested were 98.5% ± 0.4%, 98.8% ± 0.2%, 98.0% ± 1.5%, 98.9% ± 0.2%, and 98.4% ± 1.0% for (i) through (v), respectively (Fig. 3). Twelve days post-thaw a viable cell number of 27.3 ± 1.8, 24.6 ± 1.8, 31.9 ± 2.4, 29.0 ± 0.9, and 30.9 ± 2.2 million cells/mL was recorded for (i) through (v), respectively, as is shown in Figure 4. Untreated controls had 96.5% ± 0.7% viability and 15.6 ± 1.6 million cells/mL 12 days post-thaw.

Viable cell number after 12 days of culture following various cooling conditions. Samples marked 30 and 60 min 0°C chill were 2 mL samples containing 1 mL ELS in cryovials that were plunged directly into an ice bucket. All five tested conditions show significant increase in viable cell numbers over control at p = 0.002, using a two-tailed unpaired student's t-test. The initial cell concentration was 1.65 million cells/mL. All data are average of n = 5 ± SD.
Effect of chilling versus cryopreservation
Experiments were carried out to determine whether the different stresses, either chilling or cryopreservation followed by rapid thaw, have a different effect on the magnitude of cell proliferation. Figure 5 shows a representative effect from 12 days of reculture, for two separate experiments (Fig. 5, sets A and B).

Comparison between viable cell number 12 days after either simple chilling through plunging vials into an ice bucket (center, black), or cryopreservation (light gray, right) of two separate tests compared with control (darker gray, left). All tested conditions show significant improvement over control (set A, chilled *p < 0.001, cryopreserved +p < 0.003; set B, chilled p < 0.001 both sets). No significant difference was seen between samples that were chilled or cryopreserved at p < 0.01 level. Set A had an initial cell density of 1.76 million cells/mL while set B had an initial cell density of 1.5 million cells/mL. All data are average of n = 5 ± SD except set A where the control is average of n = 10, comparisons done using an unpaired student's t-test.
Figure 5, set A showed chilled and cryopreserved samples with a viable cell number of 35.1 ± 1.3 (p < 0.001 vs. control) and 32.9 ± 1.6 million cells/mL (p < 0.005 vs. control) respectively, compared with 25.6 ± 5.4 million cells/mL for the control cells.
Figure 5, set B showed chilled and cryopreserved samples with a viable cell number of 25.5 ± 2.3 (p < 0.001 vs. control) and 29.3 ± 0.5 million cells/mL (p < 0.001 vs. control) respectively, compared with 13.7 ± 1.1 million cells/mL for the control condition.
Effect of cryopreservation reagents
Cryopreservation, but not chilling, includes the additional experimental variable of exposure to cryoprotectants. Thus, the effect of chilling with and without these reagents was investigated. Furthermore, the addition of Viaspan with no chilling was studied using 45 min culture at 37°C.
At 12 days post-treatment, viable cell density was determined to be significantly improved (p < 0.001) over the control set for samples ELS chilled in both 12% DMSO and 38% Viaspan, ELS chilled in only culture medium, and ELS incubated in 38% Viaspan at 37°C or chilled in Viaspan. Samples thawed after cryopreservation showed significant improvement over control (p < 0.005) as seen in Figure 6.

Viable cell number of samples exposed to different reagents when undergoing chilling in an ice bucket or cryopreservation. All samples' viable cell number are significantly increased over the uncooled control at p < 0.001 levels except the cryopreserved sample that improved at p < 0.02 and those experiencing only DMSO or Viaspan at 37°C where no significant difference was noted. Samples experiencing Viaspan at 37°C and Viaspan when chilled in a water bath are significantly different to each other. Initial cell density was 1.76 million cells/mL. All data are average of n = 5 ± SD except the control, which is average of n = 10, and significance determined through unpaired student's t-tests. DMSO, dimethyl sulfoxide.
ELS incubated in Viaspan at 37°C showed no significant improvement in cell density over the control samples. All chilled and cryopreserved samples apart from when ELS was chilled with only DMSO were significantly better than the control (p < 0.001). For the Viaspan sets, chilled samples showed significant improvement over control samples (p < 0.001).
Time course of cell proliferation
Figure 7 shows the impact of cryopreservation on cell number over a period of 12 days post-thaw culture. Cells were initially encapsulated at a density of 1.92 ± 0.2 million cells mL−1.

Comparing temporal cell growth between samples following cryopreservation (black) and an untreated control (gray). Cryopreserved samples show a lower viable cell number for the first 7 days of culture, a consequence of damage induced during the cryopreservation cycle, however, by the end of the 12 day culture the cell count is significantly higher (p < 0.001) in the cryopreserved samples. N = 5 ± SD, p < 0.001, using a two-tailed unpaired student's t-test.
For the first 7 days of culture, the cryopreserved samples had a lower viable cell number compared to control samples. On day 8, the cryopreserved samples began to show signs of increased proliferation and overtook the control samples in terms of viable cell numbers. Viable cell numbers were 9.0 ± 1.6 million cells mL−1 for the control versus 6.9 ± 1.0 million cells mL−1 for cryopreserved samples at day 5. Viable cell numbers were 11.5 ± 1.0 million cells mL−1 for the control versus 12.8 ± 1.0 million cells mL−1 for cryopreserved samples at day 8, and 17.9 ± 2.5 million cells mL−1 for the control versus 37.3 ± 1.5 million cells mL−1 for cryopreserved samples at day 12 showing a 2.1-fold increase over the control sample by day 12 (p < 0.001).
A separate experiment considered waiting until 12 days postencapsulation to chill. After 12 days of culture, ELS had a density of 20.2 ± 1.1 million cells mL−1 at which point one set was chilled for 45 min while the other remained in culture not experiencing a change in temperature. One hundred twenty hours after treatment no significant difference was observed between the groups with control samples having a density of 74.9 ± 7.8 million cells mL−1 versus 83.2 ± 7.5 million cells mL−1 for chilled samples. Significant break-out of spheroids from the alginate was observed above around 50 million cells mL−1, which likely affected growth above this cell density.
Unencapsulated cells
A study was undertaken where a cell suspension in a cryovial was chilled and then returned to T175 flask culture. Confluence was measured for 7 days after which point the flasks became fully confluent. No significant difference was seen between a chilled sample and an unchilled control.
Cold impact on cell function and extended culture
The metabolic changes of the ELS following chilling or cryopreservation, measured by MTT assay, was significantly improved over control samples at days 5, 7, and 11 post-treatment (Fig. 8A).
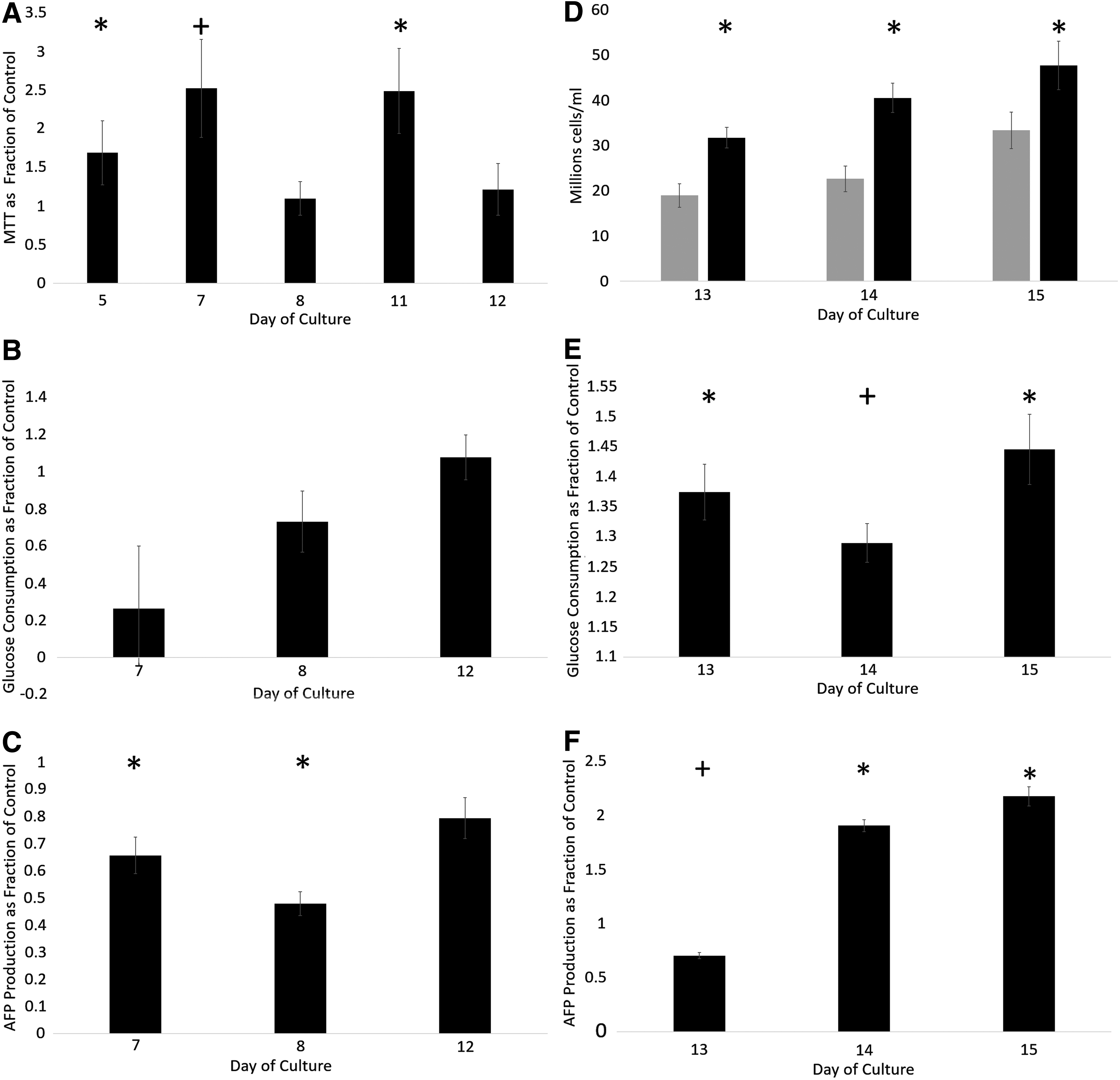
Functional assays of ELS having undergone cold treatment. Cell number for
Glucose consumption per mL biomass over 24 h is indicative of cell metabolism. There was no significant difference between the rate of glucose consumption in the ELS at any measured time point (days 7, 8, and 12) in line with their increase in cell numbers (Fig. 8B). However, AFP production per mL biomass over 24 h was significantly higher in fresh samples over cryopreserved samples.
Viable cell number and cell performance of samples examined days 13–15 post-treatment are shown in Figure 8D–F. Chilled and cryopreserved samples had significantly higher viable cell numbers at all measured time points (p < 0.001). Glucose consumption per mL ELS per 24 h (Fig. 8E) was significantly higher (p < 0.001) between days 13 and 15 for samples experiencing cold treatment over the control.
Day 13 AFP production (Fig. 8F) per mL ELS per 24 h was significantly worse in samples experiencing cold treatment over the control (p < 0.002). Samples experiencing cold treatment exhibited significantly improved performance per mL ELS per 24 at days 14 and 15 (p < 0.001).
Discussion
In this article we have demonstrated that alginate ELS that are cryopreserved or experience a reduction in temperature (≤10°C) for periods between 1 and 90 min display a greatly enhanced cell proliferation during culture 7–16 days post-treatment compared with untreated samples.
There is an extensive literature of the effects of low temperatures on mammalian cells and tissues, but we are not aware of any instances where an increase in cell proliferation was reported.
The majority of studies examining low temperature exposure on cells report negative consequences of low temperatures. The extent of these reports range from slower cell proliferation through to apoptosis and necrosis.15–19,23–26 Specific stages of the cell cycle can be disproportionately affected, 20 the G1 of the cell cycle appears particularly sensitive to the impact of low temperatures, with cells being observed to accumulate in this phase.15,18,27 The only field in which low temperatures are regularly used on mammalian tissues without freezing are in cell/tissue preservation and surgery.15,26,28,29
To our knowledge, no studies have reported the effects of cold culture temperatures to induce rapid cell proliferation; indeed, the consensus is that low temperatures are to be strenuously avoided.
We have not investigated the underlying mechanisms of this effect in this study. They may be related to cold-shock proteins, or recovery mechanisms remaining active when the initial insult of temperature reduction has passed. A previous study found no differences in alginate measured by Fourier Transform Infrared Spectroscopy between cryopreserved and noncryopreserved (or chilled) control samples. 30 The effect was not seen in cell suspensions, however, as rapid growth may have only been apparent around 7 days post-treatment, the effect may have been masked as culture beyond this point in T175 flasks results in cell detachment in the HepG2 system.
Accelerated cell growth was seen at all initial cell densities tested, varying between 1.5 and 2.13 million cells/mL. A 45-min chill test carried out at a higher cell density of 20 million cells/mL (12 days after encapsulation) did not result in increased proliferation, however, this may have been a consequence of cell break-out from the cell beads, resulting in an upper cap on ELS cell density.
Low temperature treatment has little time dependence, with samples cooled and warmed over the space of a minute still displaying increased cell proliferation compared to the uncooled control, indicating that the mechanism for inducing increased growth is triggered very rapidly. While many cell types cannot survive rapid temperature fluctuations,12,24 encapsulated HepG2 cells appear largely immune to above zero temperature fluctuations. This, combined with observed cold storage times between 1 and 90 min all inducing increased proliferation will allow this method to be used robustly in large volumes, where consistent cooling and warming times and rates are difficult to obtain across a sample. The biophysical mechanism response for this phenomenon is unclear but could be related to phase changes in membrane lipids, alteration in the cytoskeleton coupled with the unique physical environment that the cells encounter within the cross-linked alginate.
Cold treatment reduces cell number relative to the control for several days, consistent with previous literature.9,31,32 It can take up to 7 days for the cell spheroids to recover from cold-induced damage and reduction in cell numbers, and to overtake the control values. This makes the technique a potentially useful manufacturing step for systems such as bioartificial livers. The observation that cryopreserved samples display increased proliferation to the same degree as chilled-only samples is encouraging. Manufacturing a large quantity of ELS followed by cryopreservation allows for rapid delivery on demand. Cryopreservation is necessary for most bioartificial liver (BAL) or tissue-engineered constructs to be viable treatments.8,17,33
AFP production tends to be reduced during rapid proliferation. In these data, the overall performance was slightly reduced in the cryopreserved samples. As cell density was higher in the cold-experienced sample, this indicates a large reduction of around 50% in per-cell performance during the 12-day growth period. In this system, protein production tends to be inversely proportional to cell proliferation, as during the growth cycle cellular resources are directed to proliferation and not protein production. Studies examining the effects of low temperatures on hepatocytes found that when holding the cells for 30 min at 4°C, albumin production was reduced until around 7 days post-thaw, agreeing with observations in this study around the decreased AFP production. This decrease could be mitigated with addition of polyethylene glycol. 34 Albumin quantification was not undertaken in this study owing to the presence of human plasma in our culture medium, thus AFP was chosen as the indicative protein.
MTT and glucose consumption was maintained after cold treatment indicating that the cell spheroids were healthy. Viability tends to be slightly lower after cold treatment, this is due to the higher cell density making nutrient transfer more difficult. In most cases the effect was <3%, and was taken into account in viable cell numbers. Viability remained suitably high throughout, above 90% in all test conditions.
The BAL is intended for use after a 12-day initial growth period, between ∼12 and 15 days postencapsulation. 6 Cell function in this study shows increased protein production as cell growth rate slows. For effective delivery of these treatments, it would be beneficial to develop a method to arrest cell proliferation once a desired cell number is achieved to increase protein production. This would also eliminate the risk of spheroid break-out observed in 50 million cells/mL and above. Methods such as adding a small extra layer to the outside of alginate beads have been shown to stop cell proliferation when the alginate bead is full and prevent cell break-out. 3
A major consideration when developing bioartificial organs is the prolonged time required for cell culture to achieve sufficient cell number for therapeutic use. By employing low temperature treatment to ELS, we have shown that cell proliferation can be greatly upregulated, allowing a more economical cell-growth regime, reducing biomass volume required for treatment, and so substantially reducing costs. In summary, cold temperature induced proliferation could make a BAL device cheaper to culture, quicker to prepare, and more practical to deliver clinically.
Cold treatment could also allow for the possibility of very large volumes of ELS being prepared at the same encapsulation, with cryopreservation and thaw on demand and treatment commencing within 8–9 days as opposed to 26 days, which is the current setup.6,8,9,30 Exact volumes dependent on patient needs could also be determined before thaw, removing the one-size-fits-all of many BAL systems, and instead tailoring to specific patient needs without the requirement of an additional thaw.
Optimal growth has been observed in this system when the ELS have an initial cell density of 2 million cells/mL at the start of the culture period. For cell types that experience a reduced viable cell number post-thaw, it would be feasible to have a prefreeze cell density proportionally above the optimal level, which reduces to the optimum on thaw, resulting in ideal cell growth conditions.
Footnotes
Acknowledgments
Funding for this work was provided through a Medical Research Council (United Kingdom) Industrial Case Studentship (9203) and by Innovate UK (101103) between University College London and Asymptote Ltd. and a Medical Research Council Proximity to Discovery Grant (RG79366) between University of Cambridge and Asymptote Ltd.
Disclosure Statement
No competing financial interests exist.