
Abstract
A full understanding of cardiac fibroblast (cFB) biology is essential to study the adverse cardiac remodeling and recovery of myocardium infarction. However, compared to cardiac myocytes, cFBs are less well characterized. Important questions, including the variability introduced by cell age (neonatal vs. adult), culture conditions (passage, plate coating, and culture medium), and responses to stimuli (e.g., hypoxia and drug treatments), have not been well addressed and standardization of techniques is lacking. This variability invites inconsistency and the confounding of study conclusions. Thus, we here focus on characterizing cell responses and standardizing procedures for cFB isolation and culture conditions to provide reliable platforms to address important questions about cFB proliferation, activation, collagen matrix formation, and responses to relevant stimuli. Thirty litters of 1–3-day pups and 30 female (240–330 g) Sprague-Dawley rats were used to isolate neonatal and adult cFBs. We detail and validate procedures to isolate cFBs for the use of culture or direct analysis. We characterize the differences between neonatal and adult cFBs, define the changes of cFBs during serial passage, and identify the response of cFBs to different culture conditions. We have also established models for the functional screening of profibrotic and antifibrotic drugs based on cFB proliferation, myofibroblast activation, and pericellular collagen matrix formation, and models of hypoxia/reoxygenation with appropriate time course and media conditions to achieve consistent cell injury. Our standardized procedures will ensure consistency in assessing cFB function. This original contribution provides a valid platform for the ex vivo investigation of the role of cFBs in cardiac ischemia and fibrosis.
Introduction
R
The proliferation of cFBs in culture belies the complexity and difficulty of well-controlled investigations of cFBs in ex vivo experiments. Our exhaustive evaluation of the existing scientific literature shows that detailed protocols for the isolation and maintenance of cFBs in culture, especially characterization, are missing. In summary, there are at least five critical problems in this area of research. First, distinctions between neonatal and adult rat ventricular cardiomyocytes (NRVM and ARVM) are widely understood and they are selected accordingly for specific experiments. In contrast, differences between neonatal and adult cardiac fibroblasts (nFBs and aFBs) are not well understood. nFBs are more commonly used because cells are easily dissociated. Second, it is believed that the majority of nonmyocytes are cFBs. 6 Only till recently it is understood that about 20% of nonmyocytes are cFBs. 7 Then, we ask whether directly cultured nonmyocytes from enzyme digestion are pure cFBs. Third, serial passage of one to eight (P1 to P8) cFBs is commonly reported in the literature without any detailed characterization of their phenotypes.8,9 The fourth problem is the lack of quality control standards published for culturing cFBs. This is despite evidence that cFBs are environmentally sensitive.10–12 For example, it is known that mechanical cues play a role in cFB activation, 13 and the impact of culture conditions, including the stiffness of culture surfaces, has not been properly addressed. We do not know whether the autoactivation of cFBs to myofibroblasts impacts the experimental results. Last but not least, cFB studies a simple conventional culture condition that has been used to investigate cFB activation, cell proliferation, myofibroblast activation, and extracellular matrix formation. This is despite the fact that FB research is advanced and very sophisticated protocols are used to study FB of skin, lung, liver, and so on.14–16 One good example is early work on osteoblasts and skin fibroblasts showing factors such as ascorbic acid are essential for collagen synthesis in vitro.14,17,18 However, instead of direct measurement of collagen synthesis and secretion in culture, the measurement of collagen mRNA by qPCR and protein by Western blot is commonly used in the study of cFB. Close inspection of published reports reveals inconsistent data even within single reports.19,20 Clearly, we need to apply such existing knowledge to enable valid ex vivo study of cFBs. We believe that fully addressing these questions will ensure consistent and reliable experiments and allow for more valid analysis of how pathological stressors affect cFB phenotype and function.
Materials and Methods
Fibroblast isolation and cell type fractionation
This study was approved by the Institutional Animal Care and Use Committee of National University of Singapore and complied with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23, Revised 1996). Thirty litters of Sprague-Dawley neonatal rats and 30 female rats were used for cFB isolation.
aFB was isolated using a Miltenyi gentleMACS cell separation system (Miltenyi Biotec.). Heart tissue was minced to 1–2 mm, rinsed two to three times in ice-cold PBS, and then transferred to C tube for enzyme digestion (2.5 mg/mL collagenase II and 80 U/mL DNase I in HBSS, Worthington Biochemical Corp., i-DNA Biotechnology Pte Ltd). Tissue was incubated for 15–20 min at 37°C and dissociated (gentleMACS m_muscle_01 program) for three times. The contents were filtered through a 70-μm cell strainer and centrifuged at 500 g for 5 min. Cell suspension in growth medium (DMEM 10% FBS) was ready for seeding or fractionation. Additional protocols for the simultaneous isolation of nFB and NRVM, aFB and ARVM are summarized in the Supplementary Data (Supplementary Figs. S1–S3 and Supplementary Tables S1–S3; Supplementary Data are available online at
Dissociated cells were fractionated using the Neonatal Cardiac Fibroblast Isolation Kit (Rat: Miltenyi Biotec.) and magnetic-activated cell sorting (MACS). Briefly, (i) cardiomyocyte debris was removed and red blood cells (RBC) lysed using the Debris Removal and RBC lysis kits; Component 1 (Fig. 1A: C1, total nonmyocytes). (ii) The cFB population was purified by pulling-down endothelial cells and leukocytes; Component 2 (Fig. 1A: C2, endothelial cells and leukocytes). The unbound cells flowing through were Component 3 (Fig. 1A: C3, purified cFBs). Cell numbers were counted and RNA was extracted for later use.
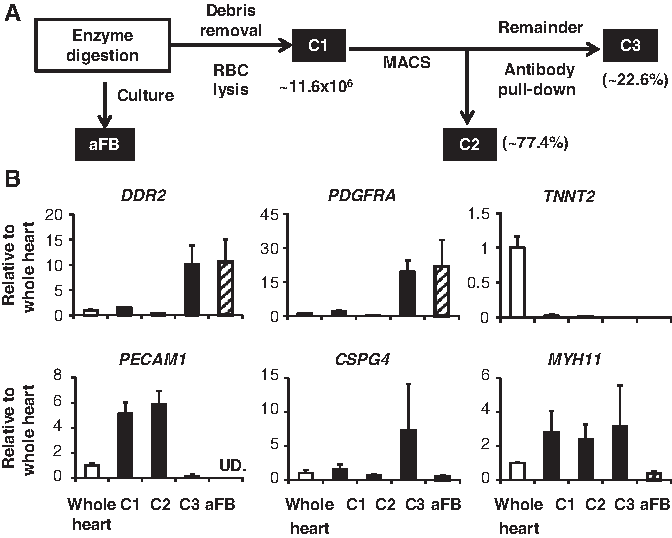
Characterization of cellular fractions from the adult rat heart and the cultured cardiac fibroblast.
Profibrotic drug treatment under different culture conditions
Both nFB and aFB were cultured in DMEM, 10% FBS. The medium was changed 2 h after seeding to remove unattached cells (P0). At 90% confluency, the cells were subcultured using 0.25% trypsin-EDTA with a 2-min incubation at 37°C (P1). Cells that did not lift up quickly were discarded. P1 cells were used in all experiments unless otherwise noted. Standard polystyrene culture surfaces were compared to soft culture surface of collagen gels (0.5 mg/mL). cFBs were serum starved with 0.25% FBS for 24 h and then treated with 100 ng/mL Ang II, 5 ng/mL TGF-β1, 10 ng/mL TNF-α, or 100 nM BNP for 24 h (aFB) or for 72 h (nFB) in serum-free DMEM. Cell numbers were counted using a MUSE cell analyzer (Merck Millipore). Cell proliferation and viability were measured by the WST-1 metabolic assay (Abcam, Singlab Technologies).
Collagen synthesis assay
FBs were serum starved for 72 h using DMEM with ITS supplement: 25 mM HEPES, 10 mg/L insulin, 5.5 mg/L transferrin, 39 nM selenium (ITS), (Gibco, Thermo Fisher), and 0.22 mM ascorbic acid where indicated (Sigma-Aldrich Corp.). Then, cells were treated with 10 ng/mL of TGF-β1 in fresh medium with ascorbic acid, which was replenished once after 72 h. After 7 days of treatment, cells and culture supernatant were collected to measure collagen synthesis and secretion.
Following debris removed by centrifugation at 600 g at 4°C and concentrated 10–20 times using Amicon® Ultra Centrifugal filters with a 50 kDa cutoff (Merck Millipore Corp.), secreted collagen in culture supernatants was quantified using the Sircol collagen assay (Biocolor, Axil Scientific Pte. Ltd.). To measure collagen content, cell lysates were subjected to native Western blots and probed with antibody against collagen type I (Sigma-Aldrich Corp.). The β-mercaptoethanol and heating steps were omitted to prevent conformational changes.
Hypoxia/reoxygenation
Hypoxia/reoxygenation (H/R) was induced by incubating cells with ≤0.2% oxygen for 8, 16, and 24 h (BioSpherix, Ltd.) and reoxygenated for 2 h under normoxic conditions. Two different buffers were used: serum-free DMEM (lg/L) or modified Esumi's buffer (137 mM NaCl, 12 mM KCl, 0.49 mM MgCl2, 0.9 mM CaCl2•2H2O, 4 mM HEPES, 20 mM sodium lactate, pH 6.2). 21 Cells were lysed for lactate dehydrogenase (LDH) activity measurement (TOX-7 assay; Sigma-Aldrich Co. LLC).
Flow cytometry and immunocytochemistry
Cells were permeabilized with 0.1% saponin, stained with primary antibodies and fluorescence-labeled secondary antibodies, and analyzed using a BD LSR Fortessa cell analyzer (BD Biosciences). For immunocytochemistry, cells were seeded onto collagen type I-coated glass coverslips, fixed in 2% neutral buffered formalin, permeabilized with 0.1% Triton X-100, and incubated with primary antibodies followed by fluorescence-labeled secondary antibodies. Cells were imaged using a Nikon Eclipse Ti (Nikon).
MiRNA stem-loop qPCR and mRNA RT-qPCR
RNA was extracted with TRIzol® Reagent (Invitrogen, Life Technologies, Thermo Fisher Scientific, Inc.) followed by DNase I treatment. MiRNA expression was determined by Stem-loop qPCR (TaqMan® MicroRNA Assay, Life Technologies) and normalized to U6B. mRNA expression of cardiac type troponin T2 (TNNT2), discoidin domain-containing receptor 2 (DDR2), platelet-derived growth factor receptor α (PDGFRα), platelet and endothelial cell adhesion molecule 1 (PECAM1), chondroitin sulfate proteoglycan 4 (CSPG4), myosin heavy chain 11 (MYH11), collagen type I and collagen type III (Col1a1, Col1a2, and Col3a), α-smooth muscle actin (ACTA2), fibronectin 1 (FN1) and its transcript variant with extra domain A (FN-EDA) were determined by universal reverse transcription and real-time PCR (iScript™ Reverse Transcription Supermix for RT-qPCR, iTaq™ Universal SYBR® Green Supermix, Bio-Rad Laboratories Pte Ltd), and normalized by TATA-box binding protein (TBP). Primers are listed in Supplementary Table S4. The ΔΔCT method was used.
Statistics
All values are presented as mean ± SEM. Data were compared for differences by one-way ANOVA followed by Bonferroni post hoc analysis or unpaired two-tail t test (GraphPad prism), as appropriate. A p value of less than 0.05 was considered statistically significant.
Results
Characterization of cell-type-specific fractions
Directly seeded cFB purity was assessed with cell-type-specific gene expression normalized to whole heart extract and compared with an alternative FB fractionation C1, C2, and C3. The fibroblast markers DDR2 22 and PDGFRα 7 were highly expressed in C3 and aFB. The MC marker TNNT2 was only detected in whole heart. The endothelial cell marker PECAM1 (CD31) 7 was highly enriched in C1 and C2 and pericyte (PC) marker CSPG4 7 in C3. Vascular smooth muscle cell (VSMC) marker Myh11 was detected in C1, C2, and C3 but greatly reduced in cultured aFB (Fig. 1B). The cell count showed that C3 (aFB) compromised < 20% and C2 (EC/WBC) about 80% of C1 (total non-MC cell), respectively.
Characterization of nFB and aFB
Gene expressions of cardiac lineage-specific and cell-type-specific markers of both nFB and aFB were measured and compared to MCs, NRVM, and ARVM. MyH6, TNNT2, and NKX2-5 were expressed only in cardiomyocytes. The cardiac transcription factor GATA4 was expressed in all four cell types but at significantly higher levels in NRVM and nFB (Fig. 2A). The fibroblast markers DDR2 and PDGFRα along with collagen type I (Col1a1 and Col1a2) were highly enriched in cFBs. nFBs generally expressed higher levels of DDR2 and COL1A1, but PDGFRα was only detected in aFB (Fig. 2A).

Characterization of the
The cardiac-enriched miR-208a and miR-133a were highly and specifically expressed in cardiomyocytes with a higher level in adult than neonatal MCs (Fig. 2B). We also identified cFB-enriched miRNAs of miR-222 5-fold, miR-214 and miR-199a-5p more than 10-fold, and miR-31 more than 50-fold higher in cFBs compared to MCs. Some miRNAs, such as miR-125a and miR-134-3p, were expressed at similar levels in MCs and cFBs (Fig. 2B).
Phenotypic shift of cFB with different passage and culture conditions
To verify molecular phenotype change, nFBs and aFBs were cultured under the same conditions (DMEM, 10% FBS) for three passages. Cell morphology changed dramatically as cells enlarged and flattened with increasing passage number (Fig. 3A, B). A gradual increase in α-SMA expression was detected from P0 to P2, along with an abrupt drop in the proliferating population as indicated by Ki67 staining. This suggests that cFBs spontaneously differentiate into a myofibroblastic phenotype in vitro. To ensure consistency and comparability, only P1 cells were used for all other experiments. The morphology of nFB and aFB, illustrated by α-SMA and vimentin immunohistochemical staining, was very similar.

nFB and aFB morphological changes over passages.
The stiffness of the culture surface plays a very important role in myofibroblast differentiation. 1 Thus, we compared the changes in cFBs cultured on type I collagen gel as a soft culture surface versus standard polystyrene surfaces. cFBs cultured on soft surface were much smaller and stress fibers stained by phalloidin were thinner, shorter, and located more to the cell periphery. cFBs on polystyrene were enlarged. Their stress fibers were thicker and more aligned across the full lengths of the cell (Fig. 4A, C). α-SMA expression was significantly decreased in soft gel cultured cells with immunofluorescent staining and flow cytometry. A notable slowdown in cFB growth was observed in collagen gel cultures and confirmed by Ki67 staining (Fig. 4B, D).

nFB and aFB phenotypic differences on collagen gel versus plastic culture surfaces. Scale bar = 50 μm. Phase-contrast microscopy and fluorescence-labeled phalloidin staining we used to visualize cell morphological and cytoskeletal features in
The response to pro- and antifibrotic drugs
The growth of cFBs was highly sensitive to the presence of serum in the culture medium (Supplementary Fig. S4A). However, cFBs showed very little response to Ang II as assessed by WST-1 assay in standard 10% FBS (Supplementary Fig. S4B). Maximum proliferative effects required 24-h serum starvation and treatment under serum-free conditions with Ang II, TGF-β1, and TNF-α with different doses (Supplementary Figs. S5 and S6). BNP had antiproliferative effects on its own and in the presence of Ang II or TGF-β1 (Supplementary Fig. S7). Both aFB and nFB showed similar responses.
The effects of drug treatment in aFBs were further tested on different culture surfaces. Phalloidin staining showed that both Ang II and TGF-β1 enhanced stress fiber development but TNF-α or BNP did not. The organization of stress fibers on soft and plastic surfaces was very different, thinner actin fibers with many cross-points versus thick and aligned stress fibers, respectively (Fig. 5A). WST-1 increased in response to Ang II and TGF-β1, and decreased to TNF-α and BNP. Similar results were observed on both culture surfaces (Fig. 5B). α-SMA mRNA expression, a test for myofibroblast differentiation, was strongly upregulated in response to Ang II and TGF-β1 and downregulated to TNF-α and BNP. The changes were much more distinct and significant on soft compared to plastic surfaces (Fig. 5C). It must be emphasized that the conventional protocols could not detect any changes in collagen or fibronectin gene expression (Fig. 5D, E, and Supplementary Fig. S8) and collagen in cell lysates by Western blot. Secreted collagen by Sircol assay was not detectable. Using our modified protocol with supplemented ascorbic acid and a prolonged 7-day stimulation, TGF-β1 treatment significantly increased cell number, α-SMA, type I collagen, and fibronectin gene expression, as well as the fibrogenic variant of fibronectin, which contains the EDA domain (Fig. 6A, B). Both collagen secretion into the medium and collagen content in cell lysates showed strong increases in response to TGF-β1 (Fig. 6C–E).

aFB responses to pro- and antifibrotic treatment on collagen gel versus plastic culture surfaces. Scale bar = 20 μm.
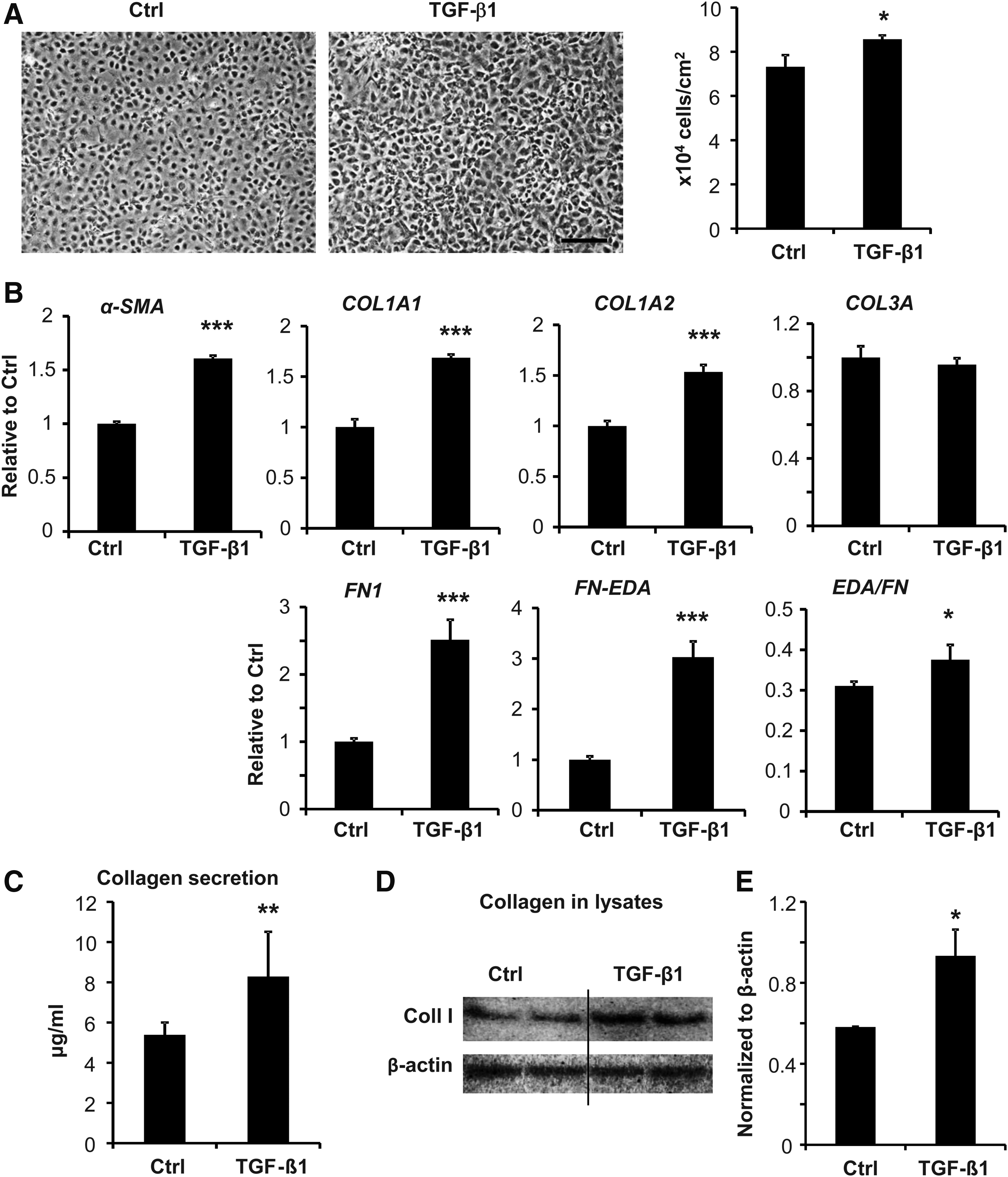
Modified protocol to detect collagen matrix formation from TGF-β-treated aFB.
The response to H/R injury
cFB H/R is a less characterized model than that of cardiomyocyte. We tested the H/R injury of nFBs in parallel with NRVM using two different hypoxia media: (i) serum-free, low-glucose DMEM (Low-Glu) and (ii) modified Esumi's buffer. NRVMs were resistant to H/R in Low-Glu that the majority of cells maintained their morphology and spontaneous beating during a 24-h hypoxia. This H/R condition induced only 20% cell loss determined by LDH (Fig. 7A, C). With modified Esumi's buffer, NRVMs quickly displayed cytoplasmic vacuoles and stopped beating at an 8-h hypoxia (Supplementary Fig. S9A). Prolonged hypoxia resulted in progressively increased cell injury. nFBs showed progressively increased levels of cell injury/death during 8, 16, and 24 h of hypoxia followed by 2 h of reoxygenation, which was similar to NRVM (Fig. 7B, D). Dramatic morphological changes were observed from the 8-h time point onward (Supplementary Fig. S9B). No LDH activity in cell lysates could be detected after 24 h of H/R. The response of aFBs to H/R was similar to nFBs (Fig. 7E).

Neonatal myocyte and cardiac fibroblast (NRVM and nFB) injury after hypoxia and reoxygenation. Scale bar = 100 μm.
Discussion
This study makes a few original contributions. We have characterized cardiac fibroblast from both adult (aFBs) and neonatal hearts (nFBs), and documented phenotypic changes with serial passage, varied culture conditions, and assorted stimuli. We have integrated existing knowledge from the noncardiac FB field to develop methods for reliable assessment of collagen synthesis and secretion. For the first time, we identified fibroblast-enriched miRNAs. Our data clearly show that no single protocol effectively allows analysis of all these aspects of cFBs in culture. As each condition possesses limitations and advantages, different conditions must be used to supplement each other both to ensure a full and accurate data set and to generate conclusions from ex vivo cFBs that recapitulate in vivo cardiac fibrosis. We have developed validated protocols for assessment of cFB proliferation, activation, and pericellular collagen matrix formation in response to pro- or antifibrotic treatment.
Due to the technical limitations and lack of specific marker, cFBs were thought to be the most populous cells in the heart for a long time. 6 The discrepancy was largely caused by the miscounting of total nonmyocyte cells, including cFB, EC, VSMC, PC, and WBC, as cFBs. 23 We used commercially available antibody-conjugated magnetic microbeads to fractionate different cell components: C1 as total nonmyocytes, C2 as EC/WBC, and the remaining C3 as cFBs and verified by cell-type-specific gene expression. There was a clear cut high expression of FB-specific genes of DDR2 and PDGFRα and undetectable MC- and EC-specific genes of TNNT2 and PECAM1 in C3. PC- and VSMC-specific genes of CSPG4 and MYH11 were still detectable in C3. A fraction of FBs was free of MC and EC but might be contaminated with PC and VSMC. cFBs consisted of ∼23% of total nonmyocytes in the normal adult heart. In this procedure, PC and VSMC were not separated, so the number is slightly higher than the previous report of ∼20%. 7
Conventionally, a mixture of isolated nonmyocytes was cultured and used as cFBs. Therefore, we asked whether nonmyocytes seeded for culture are indeed cFBs. The same panel of cell markers was measured in P1 aFB and confirmed the purity of these cells. We suspect that two key steps in the preparation are important to ensure the purity of cFB cultures. (i) Quick cell attachment of 2 h after first cell seeding is necessary to remove unattached nonfibroblasts and (ii) quick trypsinization of attached cells for subculture using a 2-min exposure with 0.25% trypsin-EDTA incubation is also critical. The differential trypsinization is a very old method based on empirical knowledge. 24 These protocols became obsolete over time along the development of new technologies such as flow- or magnetic-assisted cell sorting. However, in our case it proves to be simple, cheap, and effective. The fractionation method is effective in enriching/purifying the cFBs without subjecting them to ex vivo culture conditions. Thus, it can be very useful to assess cFB changes from an in vivo study, for example, pressure overload and myocardial infarction.
We compared gene expression of nFBs and aFBs. PDGFRα, a well-used FB marker, 25 was 100-fold higher in aFB than nFB. Similar observations have been reported in human fetal and adult cFBs. 26 PDGFα has been used as a lineage tracing marker in cFB differentiation during cardiac development. 27 It must be used with caution in a lineage tracing study. Adult and neonatal cFBs highly express vimentin and DDR2, a more specific marker for cFBs, 22 collagen type I, and collagen type III genes. Nkx2-5 is highly expressed in NRVM versus ARVM, which is consistent with its importance during early cardiac development. 28 GATA4 is highly expressed in all four of our cell preparations. For the first time, we have identified cFB-enriched miRNAs, including miR-222, miR-214, miR-199a-5p, and miR-31. Known cardiac-enriched miR-133a 29 and miR-208a were found exclusively in MCs. Some miRNAs such as miR-125a and miR-134-3p were equally expressed in MCs and cFBs. Future investigation is under way to elucidate the roles of the newly recognized cFB-enriched miRNAs in cFB biology and fibrogenesis.
α-SMA is a marker for myofibroblasts, an activated fibroblast.30,31 Although myofibroblasts are not always α-SMA positive in in vivo fibrosis,25,32 it is a good maker of FB activation in an ex vivo model. Our data indicate that α-SMA is strongly expressed in both cultured nFBs and aFBs and its expression increases with passage, a sign of spontaneous activation of FBs under conventional culture conditions. This result is consistent with other reports. 10 We also observed a decrease in cell proliferation capacity and changes in cell morphology, a sign of cell senescence, as cFB passage numbers increased. Thus, we strongly suggest that the use of early, identical passage cFBs is critically important to ensure consistency of experimental data. The spontaneous activation of normal cFBs to myofibroblasts could pose a problem in the investigation of fibrogenesis during disease progression. 1 Therefore, it is necessary to develop a culture system that could retain cFB undifferentiated phenotype.
Mechanical cues such as culture surface stiffness are important causes of the shift to a myofibroblast phenotype. 33 Normal heart tissue has a stiffness of about 10 KPa, which can increase to 70 KPa during scar formation, cardiac fibrosis, and remodeling. 34 The typical polystyrene tissue culture substrate has an elastic modulus of ∼3 GPa. 35 In effect, cFBs are typically cultured under conditions of stiffness greatly more rigid than the in vivo environment. To attenuate the factor of artificial stiffness-induced fibroblast activation, collagen gel matrices have been used as a soft culture substratum for noncardiac FBs.36,37 As it is well understood that fibroblasts from different organs are heterogeneous with respect to their physiology and morphology, 38 we need to verify this postulate to apply this knowledge in cardiovascular research to generate more physiologically relevant data in ex vivo culture experiments. Indeed, both nFBs and aFBs reduced their α-SMA expression when grown on collagen gels. Interestingly, cFB proliferation also decreased in collagen gel cultures, similar to data reported for human dermal fibroblasts. 39
Since a soft collagen gel culture offers better understanding of the reciprocal and adaptive interactions between cFB and the surrounding matrix, we further compared the effects of pro- and antifibrotic drugs on aFBs cultured on collagen gels and plastic dishes. The stress fibers were thicker and polar oriented on plastic dishes but evenly distributed with cross-points on gel culture. WST-1 assays as an indicator of cell proliferation showed similar responses to drug treatment. Pro- and antifibrotic drugs induced changes in α-SMA expression that were subtle and hard to detect in plastic cultures but became much more pronounced in collagen gel cultures. This suggests that the data obtained from in vitro models of cardiac fibrosis could be made more relevant by maintaining the stiffness of the culture surface at more physiological levels. However, profibrotic drugs could not induce any changes in collagen gene expression and protein synthesis under either culture condition.
Early work showed that factors such as ascorbic acid are critical for collagen synthesis in osteoblasts and skin fibroblasts.14,17,18 Thus, we supplemented our culture media with ascorbic acid, prolonged our treatment period to 7 days, and modified our media formulation (HEPES to maintain pH and ITS to maintain serum-free cell viability). With these modifications we observed significant increases in collagen mRNA and protein synthesis in cFB lysates and in collagen secretion into culture medium. One must be aware of the limitations and advantages of different culture conditions and to use the right protocol to address specific questions. We suggest that a conventional plastic culture is useful for a quick assessment of cFB proliferation. A soft gel culture may be needed to prevent excessive cFB activation, which may lead to the potential loss of physiological, cellular, and molecular signaling. A modified and prolonged culture condition is necessary to assess collagen matrix formation.
NRVM H/R has been widely used as an in vitro model of ischemia and ischemia/reperfusion. cFBs were suspected to be resistant to ischemic stress but little actual data tested this view.40,41 We compared two different hypoxic conditions in NRVM and nFB survival in parallel, serum-free DMEM and Esumi's buffer, an isotonic solution. 21 Surprisingly, the NRVM resisted H/R injury in serum-free DMEM, maintaining a steady number of live cells with synchronized beating despite exposure to hypoxia for up to 24 h. Both aFB and nFB cell death occurred in a time-related manner in serum-free DMEM and was not resistant to Esumi's buffer. We suggest Esumi's buffer and serum-free DMEM, respectively, for H/R studies in NRVM and cFBs.
Conclusions
In summary, cultured cFBs provide an essential ex vivo model for the study of cardiac repair and remodeling. Due to the extreme sensitivity of cFBs to conditions during isolation and culture, it is crucial to standardize methods. Our data clearly show that no single protocol effectively allows analysis of all these aspects of cFBs in culture. As each condition possesses limitations and advantages, different conditions must be used to supplement each other both to ensure a full and accurate data set and to generate conclusions from ex vivo cFBs that recapitulate in vivo cardiac fibrosis. In this study, we characterize the differences between neonatal and adult FBs and document phenotypic changes induced by serial passage and changes in culture conditions. We have established reliable and reproducible protocols for the isolation and purification of cFB for further culture or direct assessment; measurement of cFB proliferation, activation, and pericellular collagen matrix formation; and an ex vivo ischemia model. It is worth mentioning that the conventional direct-seeding method could obtain pure cFBs compared to the method with additional fractionation. It is highly recommended since this procedure is more time and cost effective. These standardized and validated protocols are useful tools for the conduct of in vitro research into the mechanisms of cardiac fibrosis and the screening of antifibrotic drugs.
Footnotes
Acknowledgments
We thank Ms. Lim Eng Siew for technical assistance in cell culture and immunofluorescence staining. We acknowledge the constructive suggestions of Dr. Paul Wolkowicz in the preparation of this article. This work was supported by grants from the Cardiovascular Research Institute Start-up Fund (National University of Singapore, Singapore) and TCR Flagship Program (P.I. A. Mark Richards, National Medical Research Council, Singapore).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.