
Abstract
Activated platelet-rich plasma (PRP) has been studied as a replacement for fetal bovine serum (FBS) in stem cell culture. However, current methods are time-consuming or require addition of exogenous substances to activate PRP, which have disadvantages in clinical applications. In this study, we developed a new method for PRP activation using a bead mill homogenizer and compared it with previous methods of PRP activation. PRP was prepared via a two-step centrifugation process and activated via calcium (Ca-PRP), freeze–thaw cycles (FT-PRP), or bead mill homogenizer processing (BM-PRP). Quantification of growth factors in PRP revealed that all forms of activated PRP released higher levels of platelet-derived growth factor-AB and transforming growth factor-β1 than those in platelet-poor plasma; however, BM-PRP resulted in significantly higher levels of growth factors than those from Ca-PRP and FT-PRP. Next, we analyzed the ability of the various forms of PRP to stimulate proliferation, migration, and differentiation of umbilical cord blood-derived mesenchymal stem cells (UCB-MSCs). Our results showed that BM-PRP significantly increased proliferation and migration rates of UCB-MSCs while maintaining the phenotypical properties and stem cell abilities of MSCs. Therefore, the developed method could be suitable for PRP activation, and the BM-activated PRP could be an adequate replacement for FBS in stem cell culture.
Introduction
S
The conventional media used for culturing MSCs generally contain xenogenic supplements such as fetal bovine serum (FBS).3,4 However, leftover FBS in cultured cells may lead to negative effects during stem cell therapy by promoting adverse immunological reactions due to the presence of xenogenic proteins or uncharacterized cytotoxic factors.4–7 For this reason, researchers have made efforts to replace animal serum with human-originated alternatives.3,7 In this regard, human blood-derived platelet-rich plasma (PRP) has been considered a good candidate to replace FBS.3,4,7,8
Many studies have confirmed that PRP enhances proliferation of various cell types in vitro, including stem cells such as MSCs.8–10 The primary factors associated with platelets are platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β, which are responsible for the proliferation of a variety of mesenchymal cells. After PRP activation, platelets secrete cytokines and their α-granule contents, and the quantity of released primary factors from the platelets varies based on the PRP activation method.11–13 Several methods to activate PRP have been developed, with only two procedures being commonly used.
About 85% of studies on MSCs with PRP conduct a freeze–thaw process to activate PRP. 9 A previous study suggested that the internal contents of PRP can be released by mechanical stress; hence, repeated freeze–thaw actions have been applied to damage platelet membranes for efficient release of growth factors into the plasma.14–16 The freeze–thaw method is safe for clinical applications because the addition of substances is not required, and thus the contents of the sample are not altered. 12 However, this method is less efficient for PRP activation than suspending the platelet lysate in saline solution because the plasma in PRP has a platelet cryopreservation effect, thus protecting the platelet from extensive damage during exposure to freezing temperatures. Accordingly, freezing is useful for long-term preservation of PRP but not ideal for PRP activation due to the lengthy time required to produce adequate platelet damage.
Other studies use calcium, thrombin, or both to activate platelets.12,17,18 These substances act on the extracellular receptors of platelets, which results in platelet aggregation and intracellular α-granule lysis. 13 These biochemical methods have been mainly used in clinical practice due to the simplicity of the procedure. However, the addition of exogenous calcium or thrombin poses health risks, including allergic reactions. 12 In addition, thrombin is mainly acquired by bovine, and immunologic reactions against this xenogenic protein have been reported to be present in many recipients19–21 ; thus, due to safety concerns, the use of thrombin is now avoided.
Because of the said drawbacks of the two commonly used PRP activation methods, there is a demand for more advanced, efficient, and safe PRP activation methods for stem cell therapy. In this regard, we developed a new method for the activation of PRP using a bead mill homogenizer (BM-PRP). The objective of the present study was to establish a more effective method for PRP activation and to evaluate the potential of BM-PRP for culturing MSCs.
Materials and Methods
PRP preparation
Umbilical cord blood (UCB) donated from ALLCORD (Seoul Metropolitan Government Public Cord Blood Bank) was used to prepare PRP. Platelet concentrations in whole blood and PRP were counted by using a hematology analyzer (BC-3600; Mindray). PRP was prepared by following a previously published protocol with some modifications. 8 First, the blood was separated using soft centrifugation (250 g for 10 min). Then, the plasma above the buffy coat was collected while avoiding contamination of red blood cells (RBC) and white blood cells (WBC). Next, the platelets from the collected plasma were pelleted by hard centrifugation (1000 g for 10 min). The platelets were resuspended with an appropriate amount of platelet-poor plasma (PPP) to standardize PRP to a concentration of 106 platelets/μL (Fig. 1A).
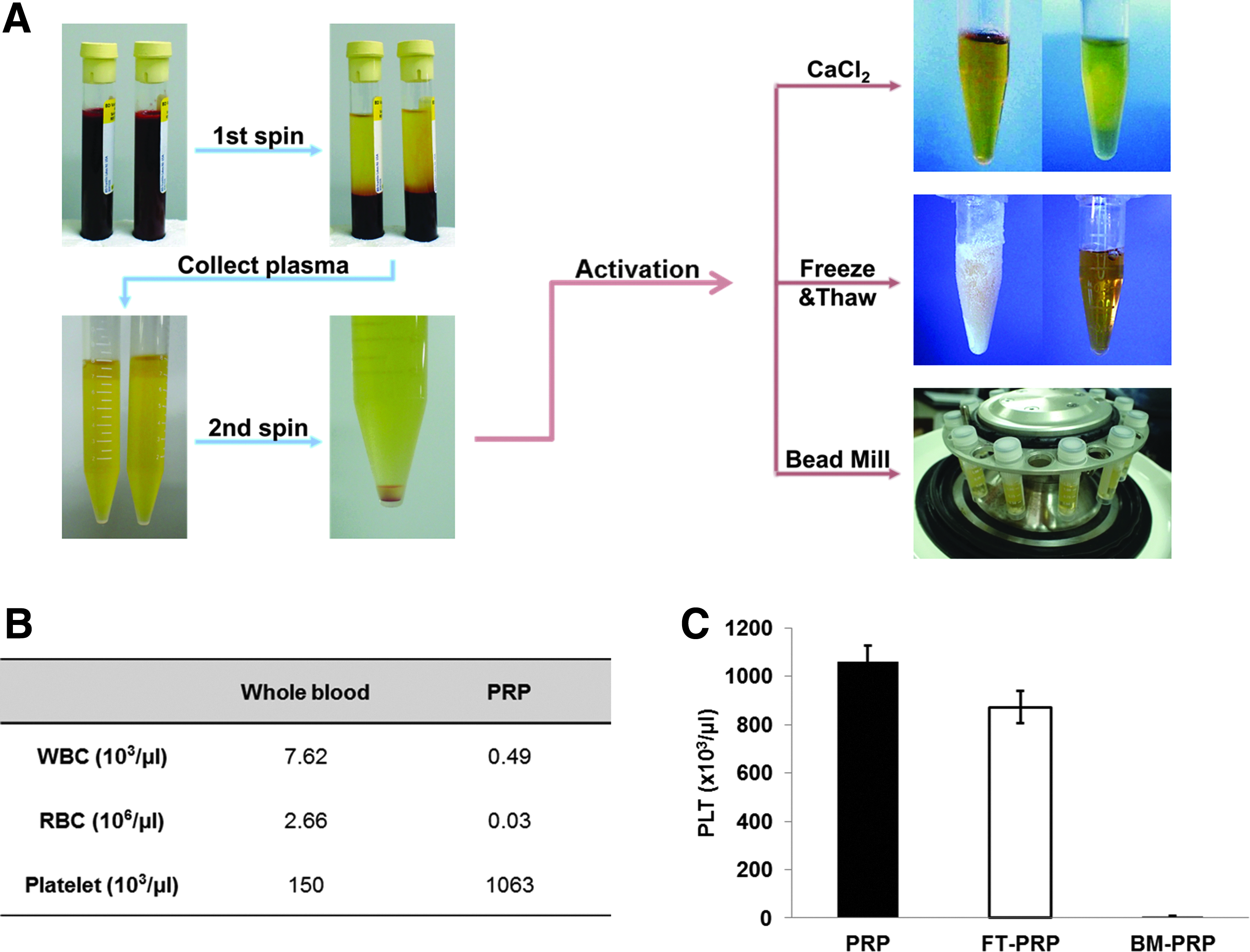
Preparation and activation of PRP.
PRP activation
PRP was activated via three methods: addition of calcium chloride, freeze–thaw cycles, and bead mill homogenizer processing. Detailed procedures are described below and illustrated in Figure 1A.
Calcium activation
Two-hundred microliters of calcium chloride solution (42.5 mg/mL of CaCl2·2H2O) was added to 2.3 mL PRP, producing a final CaCl2 concentration of 3.4 mg/mL (23 mM). The sample was incubated at 37°C (water bath) for 1 h. 22
Freeze–thaw activation
PRP activation was performed with three freeze–thaw cycles by freezing PRP at −20°C for 2 h and then thawing at room temperature for 30 min. 16
Bead mill homogenizer activation
One milliliter of PRP was aliquoted into each homogenizer tube with grinding beads. PRP was activated by bead mill homogenizer (Precellys 24; OMNI International) with three cycles at 6000 rpm for 20 s.
Platelets from the PRP samples activated via the freeze–thaw cycles and bead mill homogenizer activation methods were counted. All treated PRP samples were centrifuged at 12,000 g for 20 min at room temperature to precipitate cellular debris, fibrin clot, and platelet body. The supernatants were collected and filtered with a 0.2-μm filter to furnish the activated PRP releasate.
Growth factors analysis
PRPs were prepared from UCB units of individual donor (n = 4) and kept separately. The concentration of growth factors, PDGF-AB and TGF-β1, released by the three activated PRP samples (Ca-activated PRP [Ca-PRP], freeze–thaw-processed PRP [FT-PRP], and bead mill homogenized PRP [BM-PRP]) and PPP were determined by using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D system), according to the manufacturer's instructions. The absorbance of samples was measured at 450 nm with a microplate reader (Multiskan; Thermo Scientific). The all subsequent experiments were performed using polled PRP.
Cell proliferation
To assess the proliferative properties of the PRP products, UCB-MSCs were seeded in 24-well plates at a density of 1 × 104 cells per well with the various PRP-supplemented media. The media were supplemented with 10% Ca-PRP, FT-PRP, BM-PRP, or PPP in serum free media (MEM-alpha; Gibco) with 1% antibiotics; the same experiment with 10% FBS-supplemented medium was set up for standard comparison. The culture plates were incubated at 37°C with 5% CO2 for 72 h. The cells in each well were counted by an automatic cell counter (ADAM; NanoEnTek). The proliferation experiments were performed in triplicate (n = 3).
Cell migration
To evaluate the capacity of the PRPs in inducing migration of MSCs, we performed a transwell migration assay as described in previous reports with some modifications.23,24 UCB-MSCs (1 × 104 cells/well) were seeded with 250 μL serum-free media on the upper chamber of an 8-mm transwell membrane. Next, 750 μL 10% Ca-PRP, FT-PRP, BM-PRP, or PPP were added to the lower chamber of each transwell; the same experiment with 10% FBS-supplemented medium was used as standard. The transwell plates were incubated at 37°C with 5% CO2 for 24 h. The migrated cells were fixed using 4% paraformaldehyde and stained with 0.5% crystal violet for 5 min. The upper side of the membrane was cleaned with a cotton swab, and the crystal violet was solubilized in 10% acetic acid on a shaker for 20 min. Absorbance of the extracts was measured at 590 nm on a microplate reader. The migration experiment was conducted in triplicate (n = 3).
In vitro differentiation
UCB-MSCs were differentiated as described previously with some modifications. 7
For differentiation into osteogenic cells, UCB-MSCs were seeded at 3 × 104 cells/well in six-well plates with FBS, Ca-PRP, FT-PRP, or BM-PRP as the primary culture media. After 3 days of incubation, the cells were washed in phosphate-buffered saline (PBS) and then cultured for 10 days in Dulbecco's modified Eagle's medium (WELGENE) supplemented with 0.1 μM dexamethasone, 100 μM
For differentiation into adipogenic cells, UCB-MSCs were seeded at 6 × 104 cells/well in six-well plates with FBS, Ca-PRP, FT-PRP, or BM-PRP as the primary culture media. After 3 days of incubation, the cells were washed in PBS and then cultured for 14 days in Iscove's modified Dulbecco's medium (WELGENE) supplemented with 1 μM dexamethasone, 0.2 mM indomethacin, 0.5 mM 3-isobutyl-1-methylxanthine, and 10% FBS. Adipogenic differentiation was evaluated by using Oil Red O staining.
Flow cytometry
Cell markers were analyzed by following a previously published protocol. 7 UCB-MSCs with 10% BM-PRP or FBS-conditioned media were incubated at 37°C with 5% CO2 for 72 h. The cells were detached using trypsin, washed twice in PBS containing 1% bovine serum albumin, and then stained with anti-CD34-FITC, anti-CD44-PE, anti-CD45-FITC, anti-CD73-PE, anti-CD90-FITC, and anti-CD105-FITC (all purchased from eBioscience). Nonstained isotype cells were used as negative controls. Stained cells were analyzed by a FACS Canto II flow cytometer (BD Biosciences), and the data were analyzed using the FLOW-JO software.
Real-time PCR
UCB-MSCs with PRP or FBS, conditioned and incubated as described above, were detached, washed, and collected as cell pellets for RNA extraction. RNA was extracted by using the RNeasy Mini Kit (Qiagen), according to the manufacturer's protocol. The purity and concentration of RNA were measured via absorbance at 260 nm in a spectrophotometer (1.8–2.0). One microgram of RNA was used for cDNA synthesis with the AccuPower RT PreMix Kit (BIONEER) by following the manufacturer's instructions.
Real-time polymerase chain reaction was performed using the LightCycler 96 with SYBR Green for the detection of gene expression. Sixty-five reaction amplification cycles were performed. GAPDH was used as an internal control. The primers used are listed in Table 1.
Statistical analysis
Quantitative data are expressed as mean ± standard deviation. One-way analysis of variance (ANOVA) with Tukey's multiple comparison test was performed for comparison of each group. A p-value of <0.05 was considered statistically significant.
Results
PRP preparation
Human UCB was processed via a two-step centrifugation process to collect concentrated PRP. Counts of WBC, RBC, and platelets in whole blood and PRP are provided in Figure 1B. The platelet concentration in PRP was ∼1 × 106 platelets/μL, and that of leukocytes was 0.5 × 103 cells/μL.
Alteration of platelet number in PRP after activation
Freeze–thaw cycles and bead mill homogenizer processing are both mechanical activating methods for PRP, which work by damaging the platelet membrane. Therefore, the number of the damaged platelets can indicate the level of PRP activation. To indirectly compare the efficiency of both methods, we determined the platelet concentration of PRP after freeze–thaw cycles and bead mill homogenizer processing. Our results indicated platelet loss of morphology for 20% of platelets in FT-PRP and 99% of platelets in BM-PRP (Fig. 1C). Ca-PRP was excluded from this examination because calcium induced platelets to aggregate and the plasma to gelate, thus platelet counting was not possible.
Quantification of growth factors in PRP
To assess the activity of PRP, we measured the concentrations of PDGF-AB and TGF-β1, which are the two most abundantly produced growth factors in platelets and are essential factors in the growth of MSCs.9,15 For PDGF-AB, Ca-PRP and FT-PRP had similar concentrations, which were 8 to 10 times greater than that of PPP (p < 0.001 with Ca-PRP and FT-PRP). For TGF-β1, Ca-PRP and FT-PRP also had similar concentrations, which were four to six times greater than that of PPP (p < 0.001 with Ca-PRP and FT-PRP). However, BM-PRP showed significantly higher concentrations of both PDGF-AB and TGF-β1 than those of all the other groups (p < 0.001, for the comparison with all groups), with approximately twofold higher concentrations than those of Ca-PRP or FT-PRP (Fig. 2).
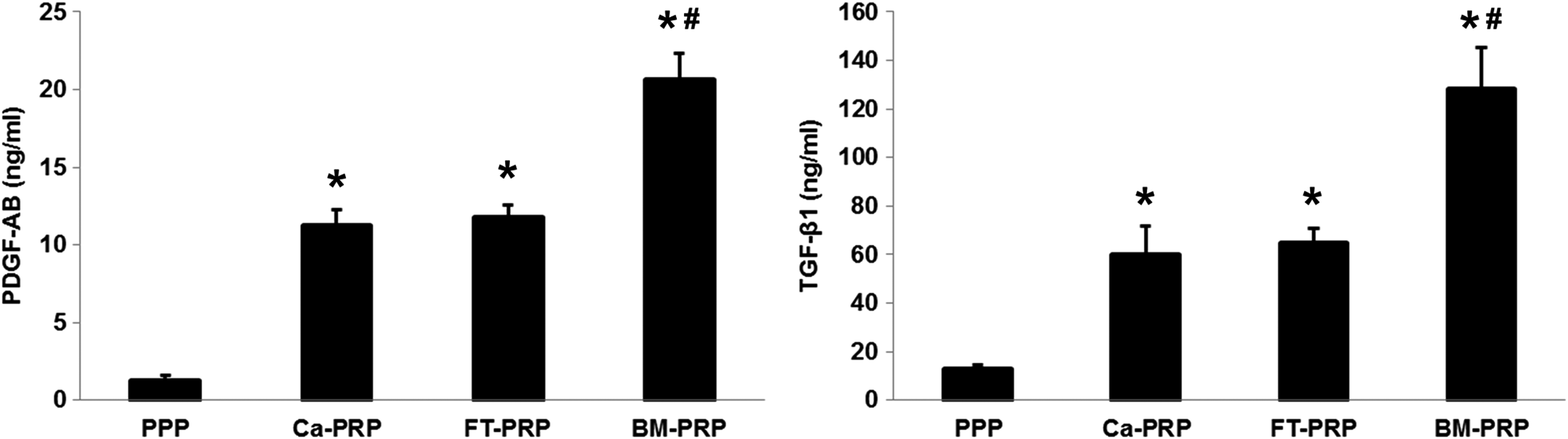
Growth factor concentrations in PPP, Ca-PRP, FT-PRP, and BM-PRP. Release of PDGF-AB and TGF-β1 was measured in PPP and the various activated PRP samples using ELISA. The amounts of released factors increased with the activation of PRP, with the significantly highest concentrations observed with PRP activation via the bead mill method (*p < 0.05 vs. PPP and #p < 0.05 vs. Ca-PRP and FT-PRP). BM-PRP, bead mill homogenized PRP; Ca-PRP, Ca-activated PRP; ELISA, enzyme-linked immunosorbent assay; FT-PRP, freeze–thaw-processed PRP; PDGF, platelet-derived growth factor; PPP, platelet-poor plasma; TGF, transforming growth factor.
Proliferation of UCB-MSCs with PRP
Given that BM-PRP contained an amount of growth factors needed to culture MSCs, we examined the proliferation of MSCs derived from UCB in response to the various activated PRPs. The proliferation rates of cells treated with PRP were significantly higher than that of cells treated with PPP (p < 0.001), and cells treated with Ca-PRP or FT-PRP caused slightly higher or similar cell proliferation compared with cells treated with FBS (no statistical differences). BM-PRP led to significantly higher levels of proliferation of MSCs than those resulting from the other PRPs or FBS (p < 0.001, for the comparison with all groups) (Fig. 3A).
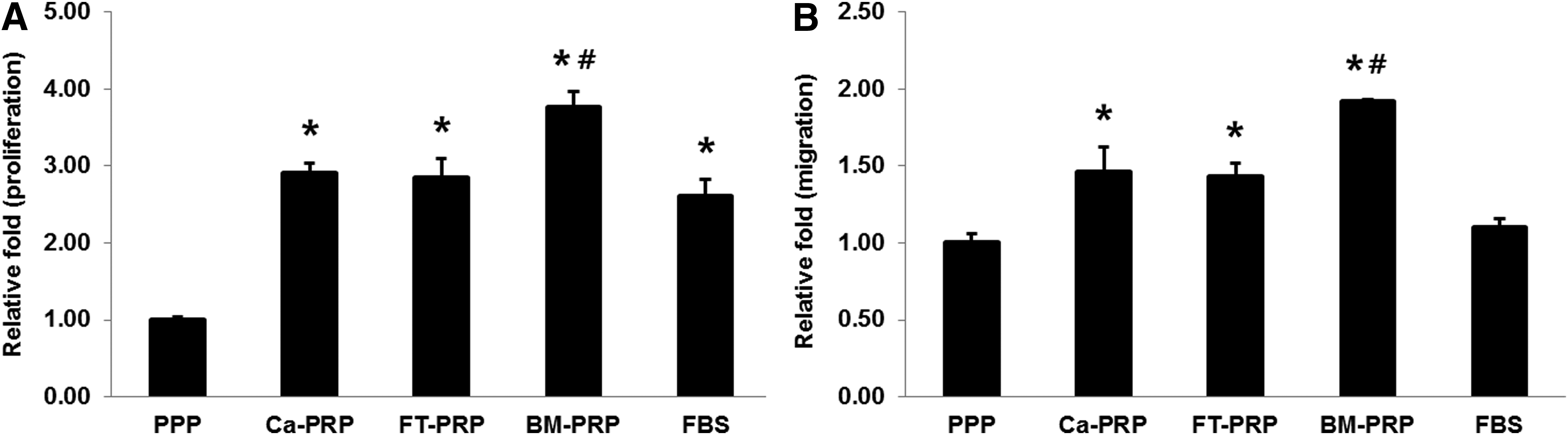
Proliferation and migration of UCB-MSCs. The proliferation
Migration of UCB-MSCs with PRP
Migration of UCB-MSCs was tested in response to the various activated PRPs. The results were expressed by relative optical density values of migrated MSCs compared with PPP as control and displayed in Figure 3B. All tested groups showed significantly higher migration capacity than that of PPP (p < 0.001 with Ca-PRP, p < 0.01 with FT-PRP, and p < 0.001 with BM-PRP), but there was no significant difference in migration capacity between PPP and FBS. There were significant increases in migration of MSCs with all activated PRP compared with that with FBS (p < 0.01 with Ca-PRP and FT-PRP and p < 0.001 with BM-PRP). BM-PRP showed the highest migration capacity among the PRP-activated groups compared with the migration capacity of Ca-PRP and FT-PRP (p < 0.001).
Differentiation of UCB-MSCs with PRP
UCB-MSCs cultured in the various media were examined for their capacity of differentiation into adipogenic and osteogenic cells (Fig. 4). In all media, MSCs successfully differentiated into adipocyte and osteoblast cells. Compared with cells cultured in FBS, those cultured with PRP showed greater differentiation capacity into osteogenic cells, especially those cultured with BM-PRP. Adipogenic differentiation was comparable in all groups.
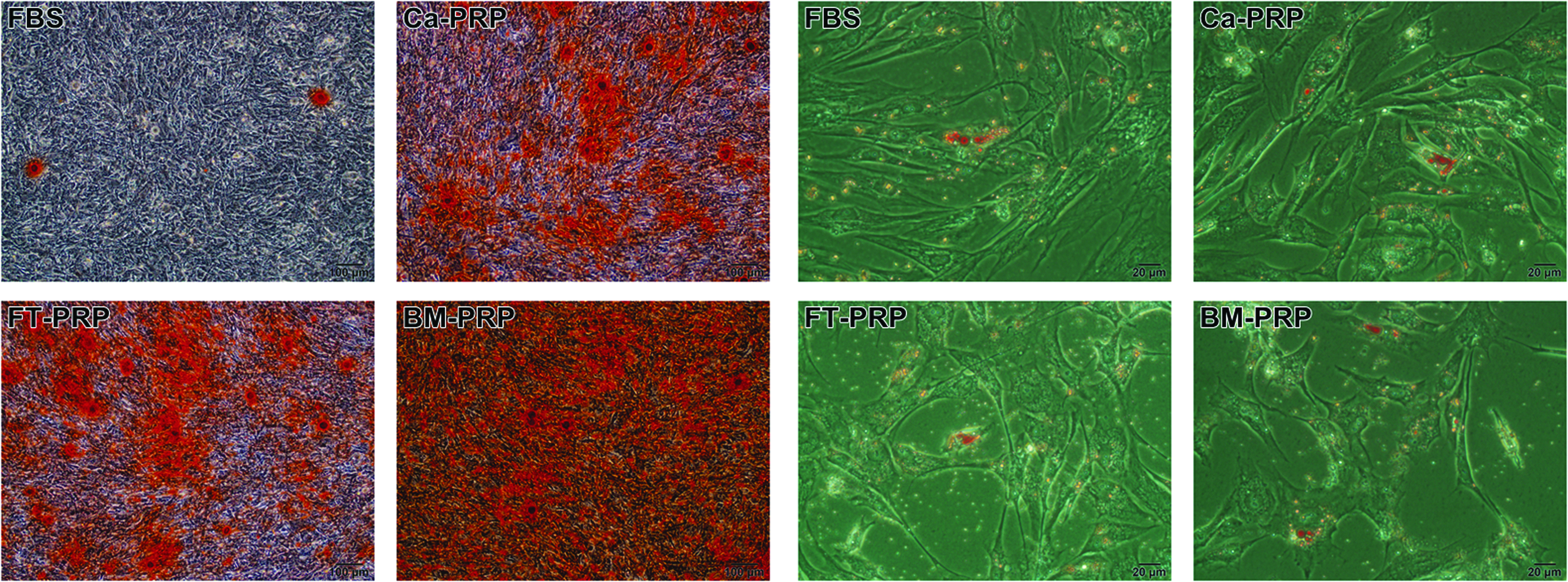
Osteogenic and adipogenic differentiation of UCB-MSCs. UCB-MSCs treated with the various activated PRPs and FBS were differentiated into osteoblasts (left: Alizarin red stain, 100 × ) and adipocytes (right: Oil Red O stain, 400 × ). The PRP treatments induced an increased differentiation capacity into osteogenic cells; in particular, BM-PRP showed the greatest differentiation effect. No significant differences among all groups were observed for adipogenic differentiation.
Expression of stem cell markers of UCB-MSCs cultured with BM-PRP
To ensure that MSCs hyperproliferated with BM-PRP maintain their phenotypical characteristics and function; we examined their cell surface and stemness markers.
To confirm the conservation of phenotypical characteristics, MSC-specific expression of markers in BM-PRP–cultured UCB-MSCs was evaluated using flow cytometry and compared as presented in Figure 5A. The expression of hematopoietic markers CD34 and CD45 was not observed, whereas the expression of mesenchymal markers CD44, CD73, CD90, and CD105 was observed. The expression levels of positive markers in cells were similar between FBS and BM-PRP.
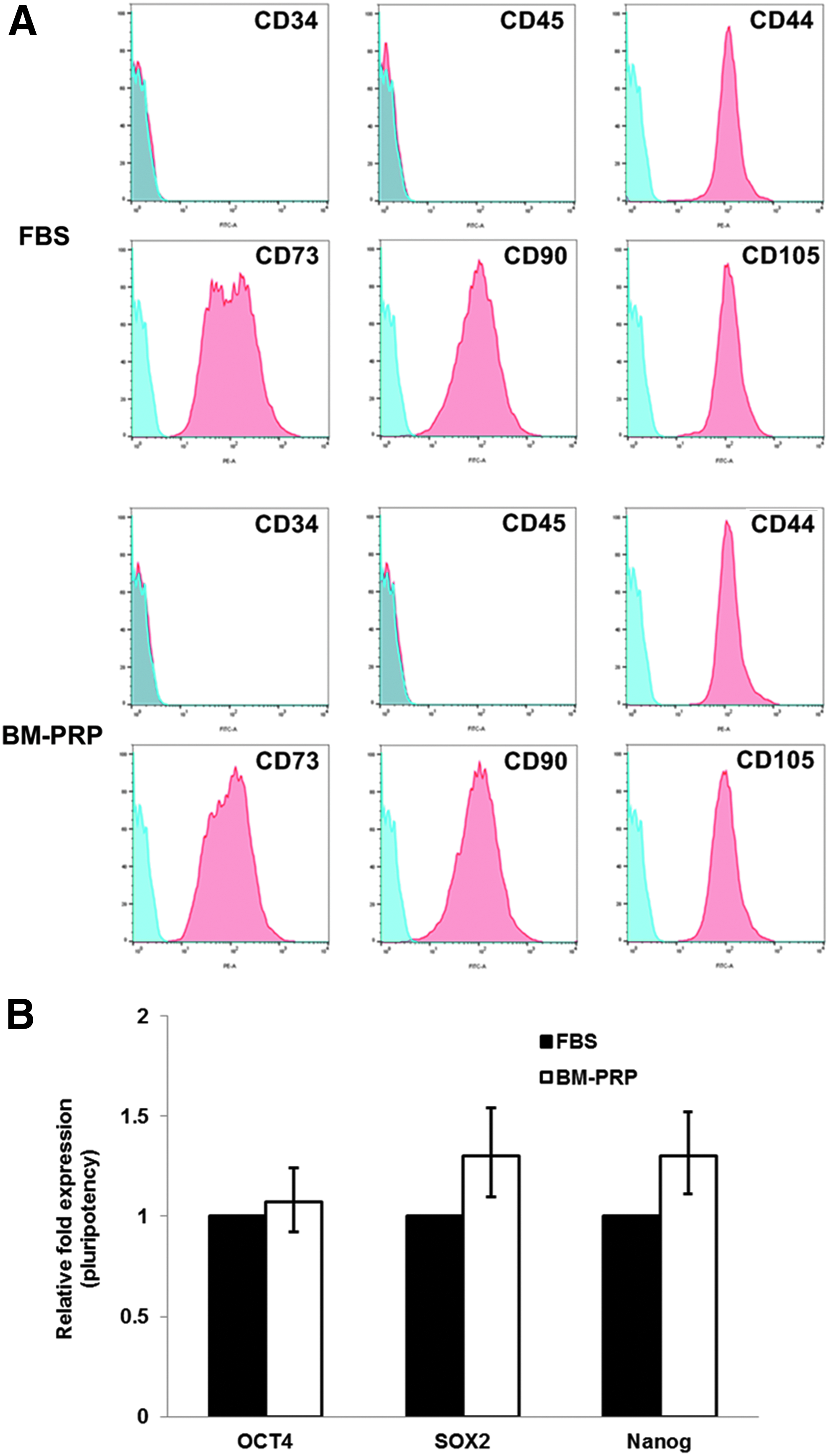
Expression of cell surface markers and pluripotency marker genes in UCB-MSCs.
To identify stemness of BM-PRP–cultured MSCs, several pluripotency marker genes were examined: OCT4, SOX2, and NANOG. Expression levels of these marker genes in BM-PRP–cultured MSCs were comparable or slightly higher than those in FBS-cultured cells (Fig. 5B).
Discussion
PRP has been widely used in various medical fields, including tissue engineering, regenerative medicine, and clinical practice, due to their abundant content of growth factors. The growth factors released from PRP are mostly influenced by its activation.11,12 However, current methods for the activation of PRP have some disadvantages, including time-consuming procedures and addition of external materials that can lead to risks of immunological side effects and cell interference.4,12
To improve the use of PRP for stem cell culture, we developed a novel method to activate PRP with a bead mill homogenizer. The use of a bead mill homogenizer produces mechanical damage that presents as rupture of the platelet membrane. The lysed platelets then release their intracellular content, including growth factors. To verify the utility of the newly developed method, we performed comparative analyses with previously used techniques, namely freeze–thaw cycles and calcium addition.
Normal platelet count in blood ranges from ∼1.5 × 105 to 3.5 × 105 platelets/μL, whereas platelet count after PRP is often found to be at least 1 × 106 platelets/μL.25,26 Generally, leukocyte-poor PRP is preferred for clinical applications, especially for cell therapy, because the presence of inflammatory cytokines may induce immunorejection by the recipient, thus leading to failure of therapy. To prevent this problem, we collected platelets with minimal concentrations of leukocytes.
To assess the efficacy of the bead mill homogenizer method for the activation of PRP, we determined the platelet concentration in FT-PRP and in BM-PRP and found significantly lower concentrations of platelets in BM-PRP than in FT-PRP. As previously mentioned, both freeze–thaw cycles and bead mill homogenizer processing mechanically activate PRP by destroying the membrane of platelets via similar mechanisms. Therefore, we expected a correlation between the decreased number of platelets after these procedures and the activity of PRP. Indeed, our experimental results suggest that the bead mill homogenizer method is more effective for the activation of PRP in exporting intracellular content than the freeze–thaw process.
To confirm the above result and to estimate the potential of BM-PRP for regenerative medicine using stem cell therapy, we examined the concentration of growth factors PDGF-AB and TGF-β1 in UCB-derived PRP and the activity of PRP on in vitro proliferation and migration of UCB-MSCs. Accordingly, we detected significantly higher concentrations of both factors in activated PRPs than in PPP. This result agrees with previous reports that show abundant amounts of PDGF-AB and TGF-β1 released from platelets after PRP activation.8,11,12,17 Ca-PRP and FT-PRP released comparable concentrations of both factors, whereas BM-PRP released a significantly higher concentration of both factors than those released by the conventional methods. Thus, the bead mill homogenizer method induced PRP to release a larger amount of growth factors than those released after the freeze–thaw process or calcium activation.
For the UCB-MSCs cultured in various activated PRPs, the higher proliferation and migration values were observed compared to those in PPP. Especially, BM-PRP treatment significantly increased both proliferation and migration of MSCs compared with treatment with Ca-PRP, FT-PRP, or FBS. This result suggests that BM-PRP released higher concentrations of the essential components necessary to support cell proliferation and migration than those released from the other PRPs or FBS. Indeed, PRP contains several growth factors that stimulate cell proliferation or migration, such as basic fibroblast growth factor, hepatocyte growth factor, insulin-like growth factor, PDGF, and TGF-β1.3,7,8,12,27 Among these, PDGF and TGF-β1 are the most abundant growth factors derived from platelets and are involved in proliferation and migration of MSCs.8,9,12,28,29
Another important factor that we evaluated was the effect of BM-PRP on multilineage differentiation of UCB-MSCs. The UCB-MSCs cultured in the various activated PRP media exhibited differentiation into mesodermal lineages, namely osteoblasts and adipocytes. Cells cultured with PRP were more easily induced for osteogenic differentiation than cells cultured with FBS, especially BM-PRP. The effects of PRP on MSCs have been reported in previous studies; in addition to proliferation stimulation, PRP triggers differentiation, especially differentiation of MSCs into osteoblasts.4,7,9,27,30–32 The factors involved in inducing differentiation of MSCs are ambiguous; however, several growth factors such as TGF-β, PDGF, and FGF are known to be involved in osteogenic differentiation.33–35 Therefore, based on our results, we infer that the abundant cytokines in BM-PRP promoted differentiation, thus proving that the BM-PRP–cultured UCB-MSCs retained their ability for multilineage differentiation.
MSCs are capable of multipotent differentiation and self-renewal. 36 Therefore, although we confirmed (through verifying multilineage differentiation) that the stemness of the PRP-cultured UCB-MSCs was maintained, the biological quality of hyperproliferation of UCB-MSCs with BM-PRP was also evaluated. Accordingly, we screened the profile of positive and negative markers by following the recommendations of the International Society for Cellular Therapy. 37 We found that stromal cell markers, but not hematopoietic markers, were expressed in MSCs cultured in either BM-PRP–or FBS-supplemented medium. These results on markers expression agree with previous studies of UCB-MSCs cultured in media supplemented with FBS, human adult blood-derived PRP, or UCB-PRP. 7,38–40 In addition, we evaluated the pluripotency of the BM-PRP–cultured MSCs by examining the expression levels of genes OCT4, SOX2, and NANOG, which are involved with self-renewal of stem cells. The expression levels of these marker genes with BM-PRP were comparable or slightly higher than those with FBS. Taken together, our results confirmed that cells cultured with BM-PRP effectively maintained their phenotypic properties (characteristics of mesenchymal cells) and pluripotency (stem cell function).
Conclusion
In conclusion, we successfully established a novel method to prepare and activate PRP. The method utilizes a bead mill homogenizer to activate PRP in an efficient and effective manner. The BM-PRP enabled the release of higher concentrations of growth factors and higher levels of proliferation and migration of UCB-MSCs than those observed with conventionally activated PRP or FBS. Furthermore, the BM-PRP–cultured UCB-MSCs conserved their phenotypical properties and stem cell-characteristic abilities, including multipotent differentiation or self-renewal. Overall, our results support that the developed method using a bead mill homogenizer is suitable for the activation of PRP and offers an adequate replacement for FBS in stem cell culture.
Footnotes
Acknowledgment
This study was supported by a grant of the Korea Institute of Radiological and Medical Sciences (KIRAMS), funded by Ministry of Science, ICT, and Future Planning, Republic of Korea (1711031808/50535-2017).
Disclosure Statement
No competing financial interests exist.