
Abstract
Advances in tissue engineering have permitted assembly of multilayered composite tissue constructs for potential applications in the treatment of combined hard and soft tissue defects and as an alternative in vitro test model to animal experimental systems. The aim of this study was to develop and characterize a novel three-dimensional combined human alveolar bone and gingival mucosal model based on primary cells isolated from the oral tissues. Bone component of the model was engineered by seeding primary human alveolar osteoblasts into a hydroxyapatite/tricalcium phosphate scaffold and culturing in a spinner bioreactor. The engineered bone was then laminated, using an adhesive tissue sealant, with tissue-engineered gingival mucosa consisting of air/liquid interface-cultured normal human gingival keratinocytes on oral fibroblast-populated collagen gel scaffold. Histological characterization revealed a structure consisting of established epithelial, connective tissue and bone layers closely comparable to normal oral tissue architecture. The mucosal component demonstrated a mature epithelium undergoing terminal differentiation similar to that characteristic of native buccal mucosa, as confirmed using cytokeratin 13 and cytokeratin 14 immunohistochemistry. Ultrastructural analysis confirmed the presence of desmosomes and hemidesmosomes in the epithelial layer, a continuous basement membrane, and newly synthesized collagen in the connective tissue layer. Quantitative polymerase chain reaction (qPCR) assessment of osteogenesis-related gene expression showed a higher expression of genes encoded collagen I (COL1) and osteonectin (ON) compared with osteocalcin (OC), osteopontin (OP), and alkaline phosphatase (ALP). Enzyme-linked immunosorbent assay quantification of COL1, ON, and OC confirmed a pattern of secretion, which paralleled the model's gene expression profile. We demonstrate in this study that, replicating the anatomical setting between oral mucosa and the underlying alveolar bone is feasible and the developed model showed characteristics similar to those of normal tissue counterparts. This trilayered model therefore offers great scope as an advanced and anatomically representative tissue-engineered alternative to animal models.
Introduction
T
Over the last few decades, there has been a substantial amount of innovation and progress in the engineering of various tissues found in the orofacial region, such as cartilage, bone, mucosa, and periodontium. 5 This has encouraged researchers to develop more intricately structured hybrid tissues that differ in the characteristics of their constituent parts, yet comprise a single functional unit. To date, only a few examples of composite tissues have been engineered, which replicate the orofacial region. Recent successes include the engineering of osteochondral components of the temporomandibular joint, which comprises both articular cartilage and subchondral bone,6,7 and the engineering of a ligamentous interface between tooth and alveolar bone to replicate the bone/periodontal ligament complex. 8
Despite these advances, the three key components of majority of orofacial tissues are that of bone, fibrous connective tissue, and an overlying epithelium. Development of an accurate alveolar bone-mucosal model therefore represents another important step in the process of achieving a clinically utilizable tissue-engineered orofacial construct. We previously reported the feasibility of tissue engineering such a model using cancer and immortal cell lines, 9 although acknowledged that patient-sourced primary cells are essential for such a model to be developed to create a more accurate representation of native tissue.
The aim of this study was to develop a novel 3D combined human alveolar bone and gingival mucosal model based on primary cells isolated from the native human oral hard and soft tissues and to characterize this model qualitatively and quantitatively, to examine whether it accurately replicates the normal tissues in terms of histology, ultrastructural appearance, differentiation, and phenotype characteristics.
Materials and Methods
Study design
Gingival biopsies and bone chips were obtained with written, informed consent from patients undergoing elective oral surgery at Charles Clifford Dental Hospital (Sheffield, UK), under appropriate ethical approval from National Research Ethics Services Committee (No. 15/LO/0116). The study included simultaneous tissue engineering of oral mucosal model (OMM) and bone model (BM), then combining both to form a composite alveolar bone mucosal model (ABMM) (Fig. 1). All materials were purchased from Sigma-Aldrich (UK) unless otherwise stated.

Schematic illustration of the preparation of OMM, BM, and ABMM. The procedure involved three main steps. First: HAOBs, NHOFs, and NHOKs were isolated from oral tissues and cultivated in monolayer culture. Second: BMs were prepared by seeding HAOBs in HA/TCP scaffold and cultured in spinner bioreactor, while OMMs were prepared from fibroblast-populated collagen gel and air/liquid interface culture oral keratinocytes. Third: combination of BM and OMM using adhesive fibrin to form ABMM. ABMM, alveolar bone mucosal model; BM, bone model; HAOBs, human alveolar osteoblasts; HA/TCP, hydroxyapatite/tricalcium phosphate; NHOFs, normal human oral fibroblasts; NHOKs, normal human oral keratinocytes; OMM, oral mucosal model.
Isolation and cultivation of primary human gingival cells
Normal human oral keratinocytes (NHOKs) and normal human oral fibroblasts (NHOFs) were isolated from gingival biopsies as previously described, with some modifications. 10 Briefly, the gingival tissues were collected and kept for 4–5 h at 4°C in a transport medium consisting of serum-free Dulbecco's modified Eagle's medium (DMEM)-GlutaMAX™ (Gibco, USA) supplemented with 100 IU:100 mg mL−1 penicillin/streptomycin (P/S) and 625 ng mL−1 fungizone. NHOKs were then enzymatically isolated from the biopsies by incubating the tissue in 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) for 14–16 h at 4°C. The epithelial layer was then scraped, cut into small pieces, and plated in 25 cm2 tissue culture flask at a density of 1.5 × 106 with an equal number of i3T3 feeder layer fibroblasts. The keratinocytes were then cultured in a humidified atmosphere of 5% CO2/95% air at 37°C in Green's medium. 11
NHOFs were isolated from the connective tissue layer by incubating in 0.05% (w/v) collagenase type I (Gibco) in DMEM-GlutaMAX containing 10% fetal bovine serum (FBS) at 37°C for 4 h. The digested tissue was then centrifuged at 200 g for 5 min, and the resultant pellet resuspended in complete DMEM (CDMEM; DMEM-GlutaMAX supplemented with 10% FBS, 625 ng/mL fungizone, and 100 IU:100 mg mL−1 P/S).
Both NHOKs and NHOFs were fed thrice a week until confluency and then used at passage 2.
Isolation and cultivation of primary human alveolar osteoblasts
Primary human alveolar osteoblasts (HAOBs) were isolated from bone chips collected in a sterile bone trap during preparation of dental implant sites.12,13 After collection in the transport medium, the osseous tissues were extensively rinsed in phosphate-buffered saline (PBS) and vortexed to remove blood components. Bone fragments were cultured as explants in 75 cm2 flask in CDMEM supplemented with 50 μg/mL L-ascorbic acid 2-phosphate (L-AA) at 37°C in a humidified atmosphere of 95% air, 5% CO2. The culture was left undisturbed for 7 days, after which the medium was replaced 2–3 times/week until the culture attained confluency, whereby cells were detached by trypsin/EDTA (0.25%), subcultured, and used in the third passage.
Engineering the OMMs
A collagen-based OMM was constructed according to the technique described by Dongari-Bagtzoglou and Kashleva. 14 A solution of 10 × DMEM 13.8 mg mL−1, FBS 8.5% (v/v), L-glutamine 2 mM, reconstitution buffer (22 mg mL−1 sodium bicarbonate and 20 mM HEPES), and 5 mg mL−1 rat-tail type I collagen (R&D Systems, UK) was prepared on ice and neutralized by 1 M sodium hydroxide to pH = 7.4.
Finally, a cell suspension of NHOFs in CDMEM at a concentration of 2 × 105 cells per model was added to the solution. The resultant fibroblast-containing collagen was transferred into tissue culture inserts (0.4 μm pore size, 30 mm diameter; Millipore) and incubated at 37°C for 2 h until it solidified. 1.5 mL CDMEM was then added inside and outside the insert. After 3 days, 1 × 106 NHOK cells (per model) suspended in 50 μL Green's medium were seeded on the gel surface and allowed to adhere for 3 h. Two microliters Green's medium was then gently added into the insert and incubated at 37°C, 5% CO2 for 4 days. When epithelial cells reached confluency, the culture was raised to air/liquid interface and fed every other day for 10 days.
Engineering the BMs
Ten sterile ceramic discs (2 × 10 mm) of hydroxyapatite/tricalcium phosphate (HA/TCP) (60%/40%) (Ceramisys, Ltd., UK) with total porosity of 78.9% were used as a scaffold. The discs were placed in 24-well plates and prewetted with CDMEM 24 h before cell seeding. 2 × 106 HOB cells suspended in 15 μL CDMEM/L-AA were then added dropwise to each scaffold. The cells were allowed to adhere for 2 h, and then BMs were completely covered with 2 mL CDMEM/L-AA and incubated overnight. After 24 h, BMs were suspended in a spinner bioreactor (Branstead Stem, UK) and spun at a rate of 30 rpm. The medium was changed every 2 days for 17 days.
Cell viability assessment using PrestoBlue live assay
The assessment of cell viability within BMs and OMMs was performed using the PrestoBlue (PB) assay (Invitrogen, USA) at day 17 before the combination of the two constructs. BMs and OMMs were separately placed in a 12-well tissue culture plate, washed with PBS, and then a mixture of 900 μL of CDMEM and 100 μL of PB reagent was added to each well. Three acellular discs and collagen gel were included as negative controls for BMs and OMMs, respectively. After a 3-h incubation at 37°C, the fluorescence values (excitation/emission: 560/590 nm) of 200 μL aliquot were measured in triplicates using spectrophotometric plate reader (Infinite® M200; TECAN, USA). For calculation, the average fluorescence values of the medium containing the scaffolds without cells were subtracted from the averaged sample readings.
Engineering the composite ABMMs
ABMMs were constructed as previously described. 9 At day 17, BM and OMM models were combined using a biocompatible fibrin-based adhesive sealant (ARTISS, Baxter, UK). In brief, BMs were retrieved and placed on a sterile culture plate containing 10 mL CDMEM. The fibrin adhesive was prepared in a prefilled syringe according to manufacturer's instructions and a thin layer of the mixed fibrinogen-thrombin sealer applied to the nonepithelial side of an OMM. The OMM was then immediately attached to the surface of a BM and held in the desired position with gentle compression for a minimum of 60 s to ensure that the adhesive material had completely set and both models were firmly adhered to each other. The ABMMs were then further cultured for 5 days, after which the analyses were performed.
Histological examination of ABMM
ABMMs were fixed in 4% paraformaldehyde (PFA) for 24 h and embedded in 2-hydroxyethyl methacrylate resin (Technovit 7100; Heraeus Kulzer) according to manufacturer's instructions. Briefly, samples were dehydrated in graded ethanol series, 75%, 95%, and 100%, for at least 2 h, and then preinfiltrated with equal parts of basic solution and 100% ethanol overnight on a rotating mixer. Infiltration was performed with an infiltration medium consisting of 100 mL basic solution and 1 g hardener-1. Finally, samples were polymerized in suitable molds with a premixed 15 mL infiltration medium and 1 mL harderner-2. Polymerization was completed over the period of 2 h at room temperature.
For routine hematoxylin and eosin (H&E) staining (Leica Microsystems), the ground block was first sectioned into 100–150 μm thickness with a cutting machine (IsoMet® 1000 precision saw; Buehler UK, Ltd., UK). The section was then adhered to a glass slide using cyanoacrylate adhesive (Loctite® glass bond UV curing, UK). The thickness was then further reduced to 30–35 μm by grinding the sections with silicon carbide papers of P800 and P1200 roughness in a grinder-polisher machine (Buehler™ Metaserv, UK).
Histological, immunofluorescent, and transmission electron microscopy examinations of OMMs
OMMs were processed for histological, immunofluorescent (IF), and transmission electron microscopy (TEM) examination at the end of culture period. Frozen sections were prepared as previously described. 15 OMMs were fixed with 4% PFA for 24 h, followed by an overnight incubation in 18% sucrose in PBS. The samples were frozen in OCT Compound (Thermofisher, UK) and sectioned to 14 μm, and sections were then either stained with H&E for examination under inverted microscope (Olympus, Japan) or were subjected to IF staining.
For IF staining, the sections were washed with PBS for 5 min, permeabilized with 0.2% triton x-100 for 5 min, and then blocked with 1% bovine serum albumin in 0.1% PBS-Tween for 1 h. The sections were then incubated overnight at 4°C with conjugated anti-Cytokeratin 13 (Abcam, UK; ab198584) and anti-Cytokeratin 14 (Abcam; ab192055) antibodies at a working dilution of 1:100. After washing, slides were mounted using DAPI (4′,6-diamidino-2-phenylindole)-containing mounting medium (Thermofisher) and viewed using IF microscopy (Zeiss, Ltd., Germany). Normal oral mucosa (NOM) was used as a positive control, while negative control was OMM stained with IgG isotype (1:100) (ebioscience, UK), followed by gout anti-rabbit secondary antibodies (1:200) (Abcam; ab150083).
For TEM analysis, 4 mm sections of OMM were fixed in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer for 2 h at 4°C and postfixed in 1% osmium tetroxide for 2 h. Tissue was dehydrated with 70%, 95%, and 100% ethanol and then embedded in resin. Samples were left for 48 h in the oven at 60°C and thereafter examined using a transmission electron microscope (FEI tecnai 12 Bio-twin, 120 Kv TEM).
Quantitative polymerase chain reaction examination
For gene expression analysis, ABMMs were snap frozen, grinded, and digested with a lysis buffer. RNA was isolated using an isolate II RNA Mini Kit (BioLine, UK) according to manufacturer's instructions. Five hundred nanograms total RNA was reverse transcribed using a high-capacity RNA to complementary DNA (cDNA) kit (Life Technologies, UK).
0.5 μL cDNA was amplified using 5 μL TaqMan gene expression master mix, 3.5 μL nuclease-free water, and 0.5 μL TaqMan gene probes (Applied Biosystems). Genes encoding the following markers were then interrogated: alkaline phosphatase (ALP), osteopontin (OP), osteonectin (ON), osteocalcin (OC), collagen I (COL1), cytokeratin 10 (CK10), and cytokeratin 13 (CK13). B-2-Microglobulin was used as a reference control gene (All Applied Biosystems, UK).
Cycling conditions were holding at 95°C for 10 min, then 95°C for 10 s, followed by 60°C for 45 s, and the cycle was repeated 40 times (QIAGEN, Germany). The threshold cycle (Ct) was normalized against the reference gene (Δ Ct) and the expression relative to it was calculated.
Enzyme-linked immunosorbent assay
Commercially available enzyme-linked immunosorbent assay (ELISA) kits for COL1, ON (R&D Systems), and OC (Abcam) were used according to manufacturer's instructions to quantify these proteins in ABMM after a 24-h incubation in serum-free tissue culture-conditioned medium. The solutions were read at an absorbance of 450 nm using a microplate reader (Infinite M200; TECAN).
Statistics
All data were presented in terms of mean ± standard deviation of three independent experiments performed in triplicate. One-way analysis of variance complemented by Tukey's multiple comparisons test was performed using GraphPad Prism v7.0 (GraphPad Software, La Jolla, CA) and differences were considered significant when p < 0.05.
Results
Viability assay
Cell viability testing using the PB assay revealed that HAOBs within the BM as well as NHOFs and NHOKs in the OMM remained vital throughout the experiment (Fig. 2).
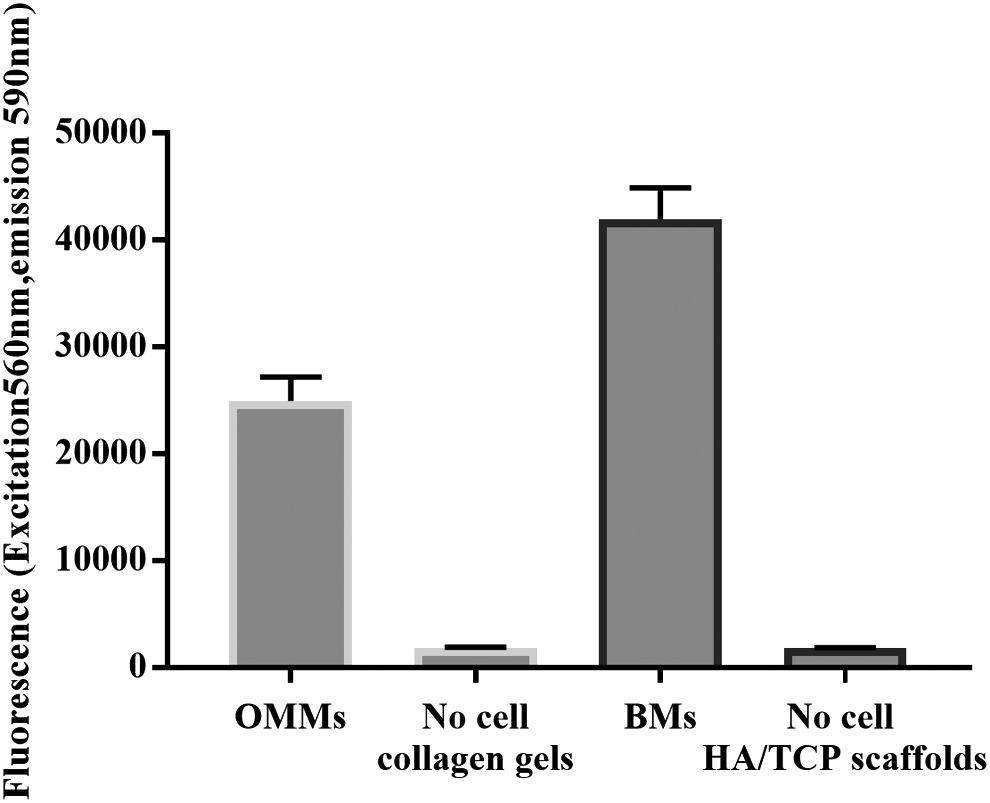
Vitality assessment of OMMs and BMs. The figure shows the activity of HAOBs within HA/TCP scaffold as well as NHOFs and NHOKs in OMMs after 17 days of culture.
Histological assessment of ABMM
Histological observation revealed that the ABMM had a structure consisting of epithelial, connective tissue and bony layers, which were comparable to the histological architecture of the soft and hard tissues of the oral cavity (Fig. 3A). The model's surface displayed a continuous stratified epithelial layer and a connective tissue layer densely populated with viable fibroblasts (Fig. 3B). The hard/soft tissue interface showed a thin band of cell-infiltrated sealant adhering both layers (Fig. 3C). Viable cells evenly populated the scaffold porosities with a secreted matrix partially or completely filling the pores of the scaffold (Fig. 3D).
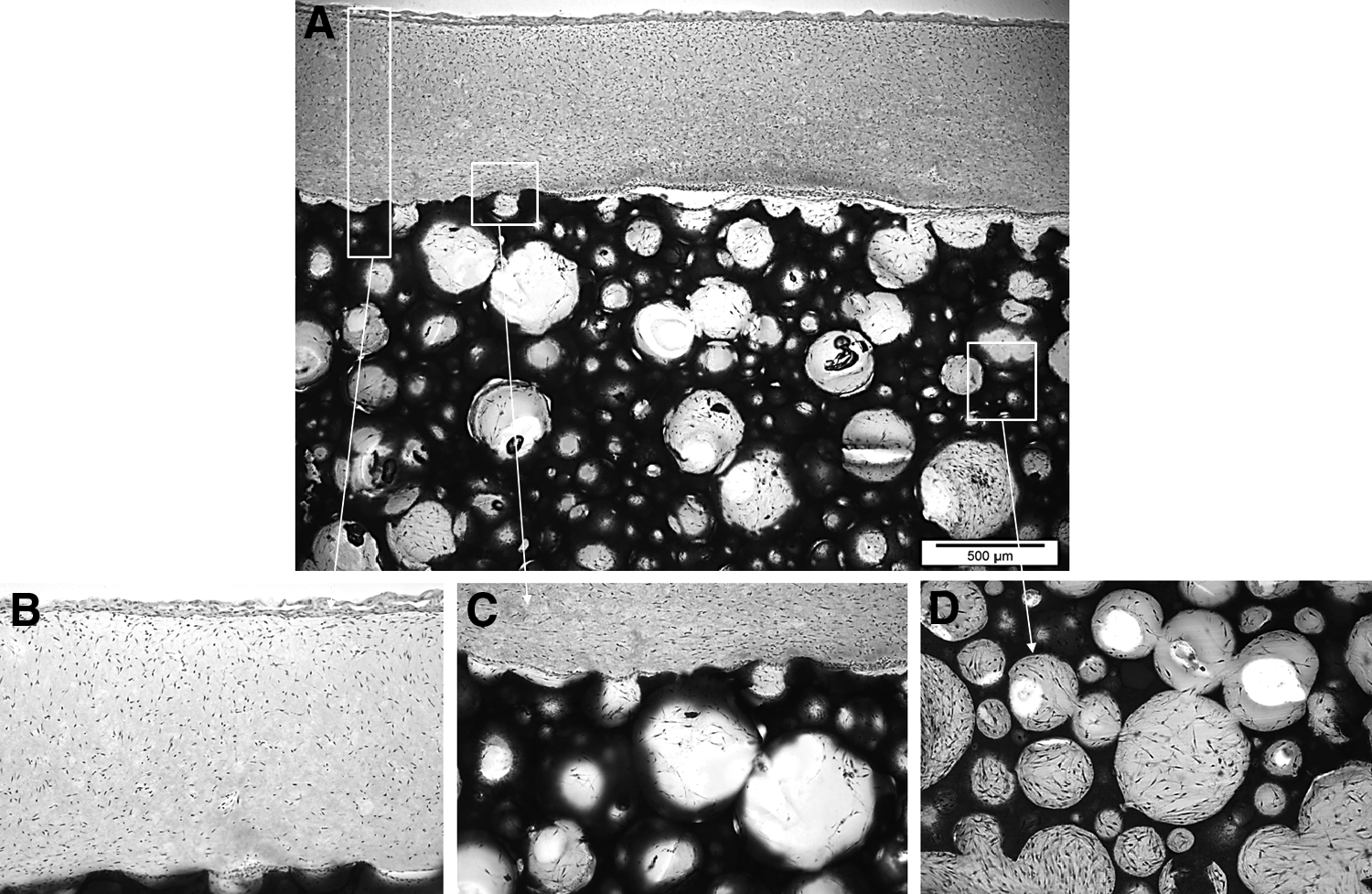
H&E-stained histological sections of ABMM showing
OMM assessments
Histologically, the OMM showed a proliferating basal layer and well-differentiated stratified squamous oral epithelium of 12–14 NHOKs thickness, which mimicked that of NOM (Fig. 4). The epithelium consisted of four distinct layers that included equivalents to basal, spinous, intermediate, and superficial cells, respectively. The uppermost aspect of the superficial layer had cells of a flattened appearance, while cells in the basal layer remained rounded. Glycogen granules were occasionally observed in the intermediate layer. NHOFs were found dispersed homogeneously in the connective tissue.
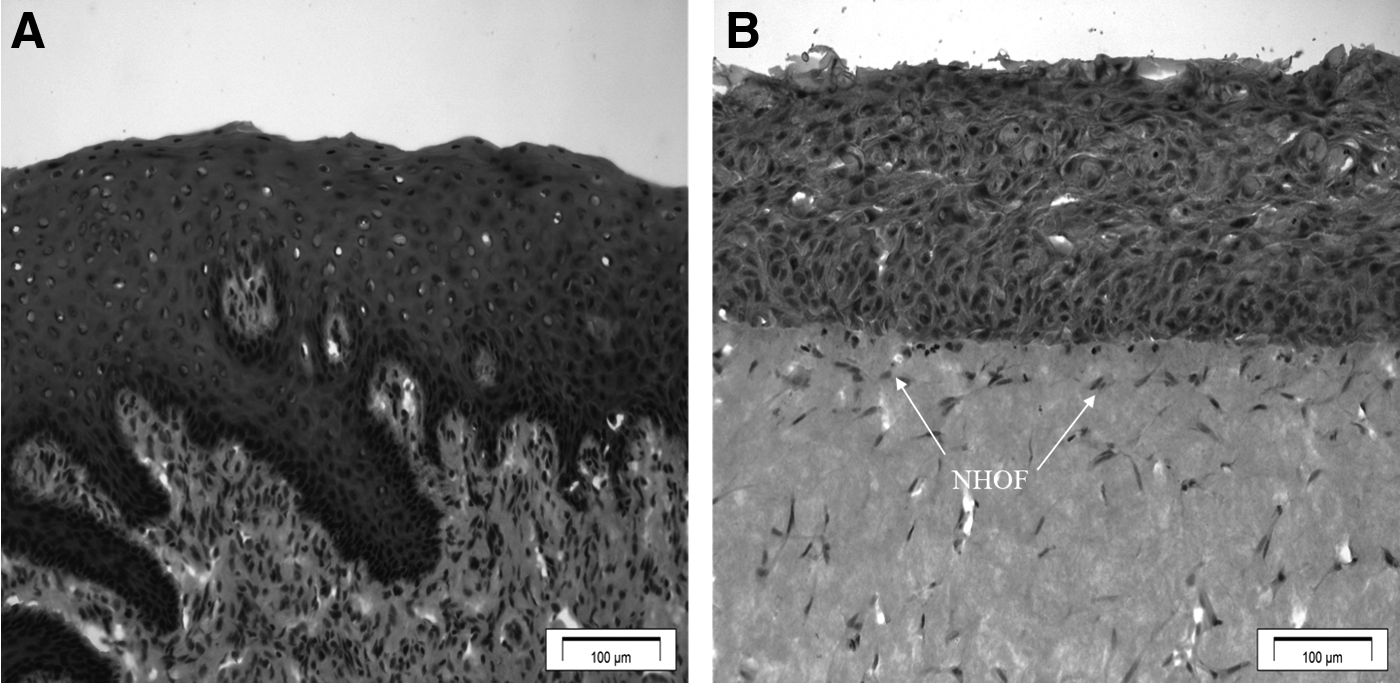
H&E-stained histological sections of
Figure 5 shows the keratin expression of NOM and OMM, as assessed by immunofluorescent staining for CK13 and CK14. The suprabasal cells of the OMM strongly expressed CK13, consistent with the staining of intermediate and superficial cells in para-keratinized stratified epithelium. 16 CK14 was expressed throughout the entire epithelium.
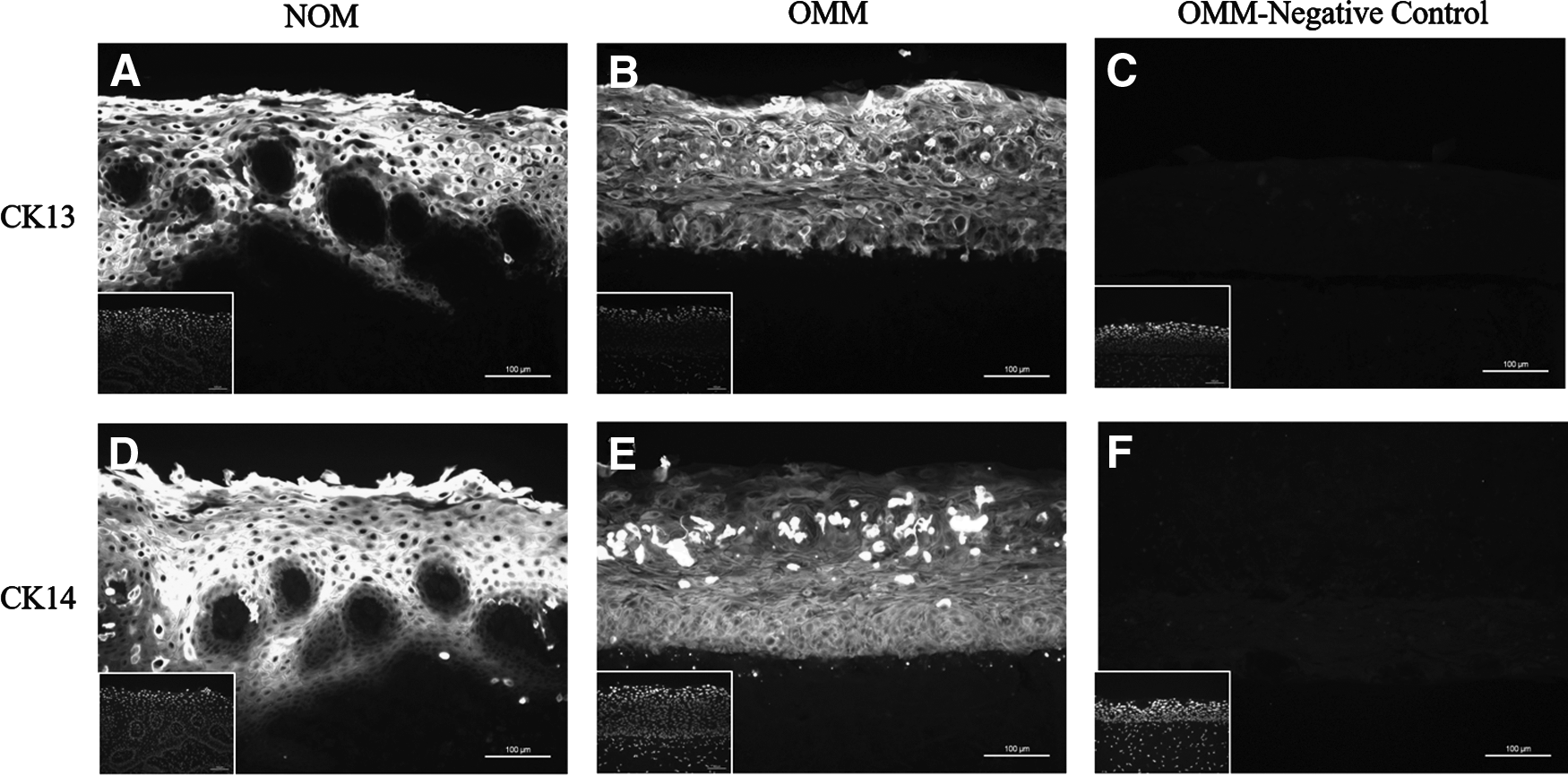
Immunofluorescent staining of the oral mucosa showing
Ultrastructural analysis of the OMM demonstrated the presence of numerous desmosomes between adjacent epithelial cells (Fig. 6A). A continuous and intact basement membrane was formed, anchoring the epithelium firmly to the connective tissue by means of hemidesmosomal attachments (Fig. 6B). In the sup-epithelial layer, a high amount of newly synthesized collagen was observed (Fig. 6C).

Ultrastructural analysis of the OMM by transmission electron microscopy showing
Quantitative polymerase chain reaction assessment
Figure 7 summarizes gene expression for the composite model, including both HAOB and NHOK cell components. Of all genes evaluated, COL1 demonstrated the highest level of expression. Bone-specific genes such as ALP, OC, ON, and OP were detected, with ON expressed in high quantities. Genes encoding CK10 and CK13 were expressed and were not significantly different.
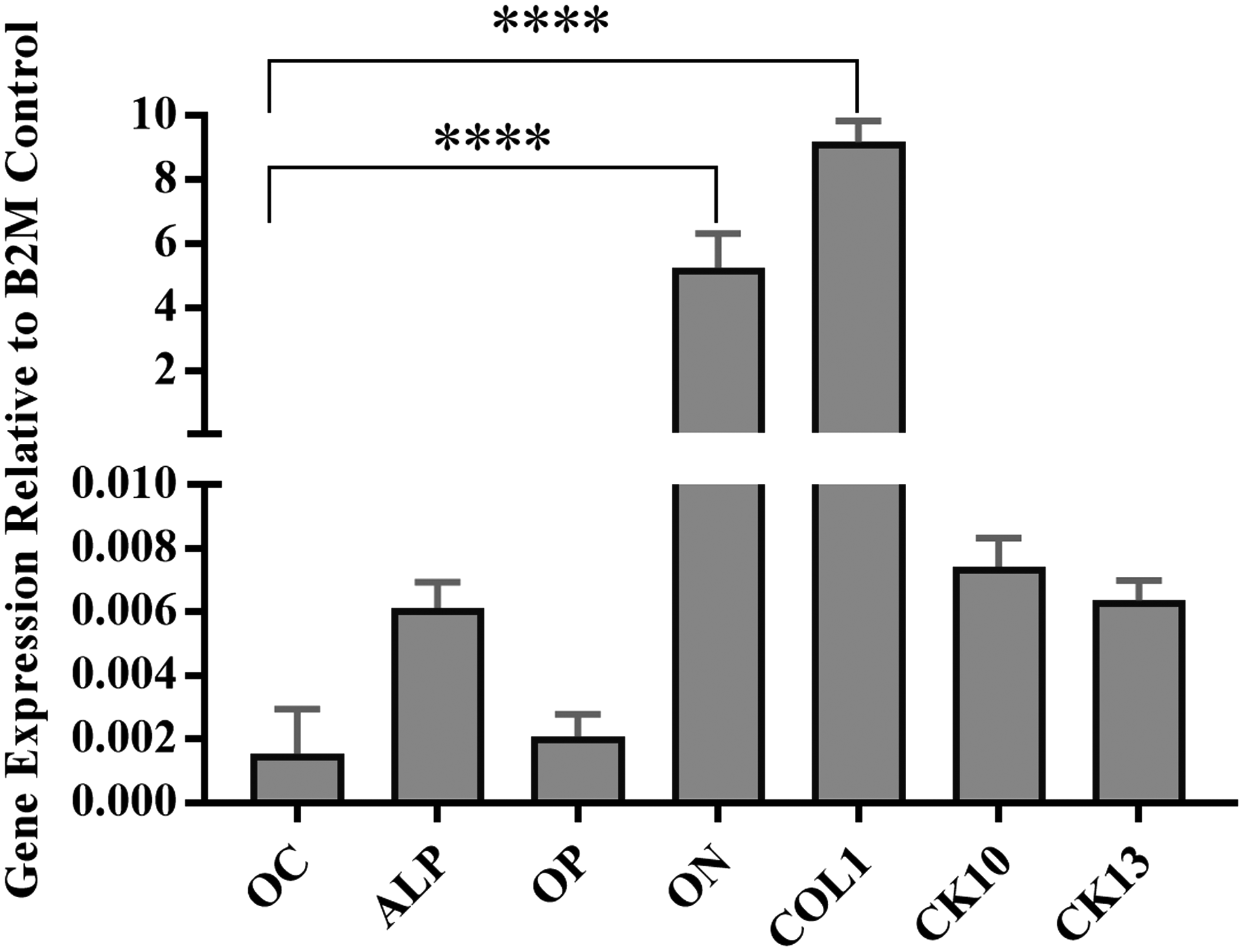
The gene expression of HAOBs and NHOKs in the ABMM as evaluated with polymerase chain reaction analysis. The osteogenic markers OC, ALP, OP, ON, and COL1, in addition to the epithelial markers CK1 and CK13, were detected. Statistical significance was determined as p < 0.05 from control (****p < 0.0001). ALP, alkaline phosphatase; OC, osteocalcin; ON, osteonectin; OP, osteopontin.
Enzyme-linked immunosorbent assay
ELISA results for COL1, ON, and OC were consistent with the quantitative polymerase chain reaction findings. COL1 demonstrated a significantly increased protein concentration in comparison to ON and OC. ON, in turn, was higher than OC (Fig. 8).
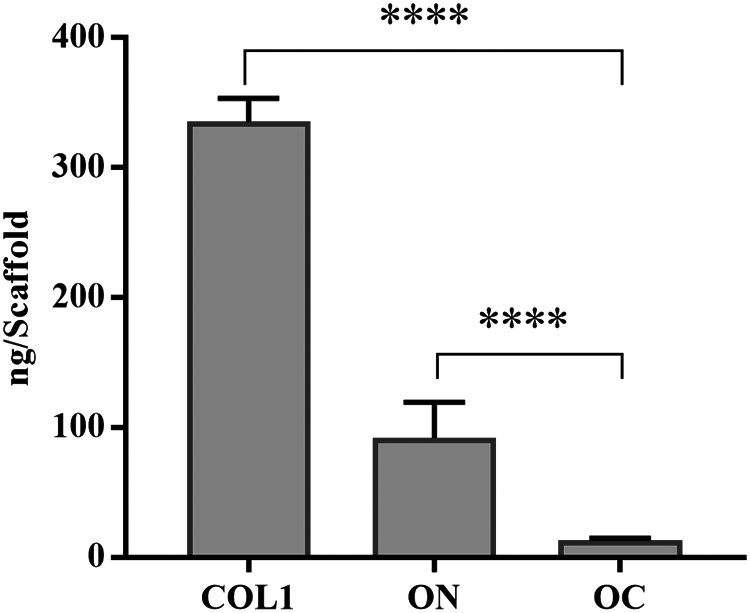
Protein expression of COL1, ON, and OC in ABMM analyzed by ELISA. Statistical significance was determined as p < 0.05 (****p < 0.0001). ELISA, enzyme-linked immunosorbent assay.
Discussion
There is an increasingly recognized need to engineer tissue equivalents for both clinical and experimental applications. In this study, we have developed and characterized an in vitro 3D human alveolar osteomucosal construct that replicates the natural anatomical relationship of oral mucosal tissue and the underlying bone.
In 3D culture, cell viability is influenced by several factors, including the scaffold and culture environment. Calcium phosphate and collagen constitute the main elements of the extracellular matrix of bone and oral mucosa, respectively. 17 Therefore, due to their biocompatibility, they have been widely used in tissue engineering as scaffolds to support cell growth without compromising its vitality. 18 The culture environment, on the other hand, that promotes mass transfer and nutrient delivery is another important factor to maintain cell vitality. In our study, growing OMM in a static culture raised no problem due to the hydrophilic, high water-containing networks of collagen hydrogel that enhance permeability for oxygen, nutrients, and water-soluble metabolites. 19 Ceramic-based BM, by contrast, required a dynamic flow to improve nutrient and waste diffusion because the static culture is only sufficient to nourish the thin superficial layer, ∼100–200 μm, contacting the medium. 20 As the cells increase in number, so does their metabolic demand and the buildup of waste products. Consequently, the deeper cells in the tissue interior can be deprived of oxygen and nutrient source in long-term static culture conditions. For this reason, the total culture time of the composite tissue after combining the hard and soft tissue constructs was limited to an additional 5 days at the static air/liquid interface in this study.
We have shown that the histological structure and expression of key markers of epithelial differentiation in OMM were comparable to those of its normal tissue counterpart. OMM displayed the characteristics of a para-keratinized epithelium, which suggests that the keratinocytes used in this model retain the para-keratinized status of the original oral mucosa from which they were derived. 21
The control of keratinocyte in proliferation and differentiation is multifactorial. Several studies have confirmed the role of fibroblasts in epithelial development through the stimulation of keratinocyte proliferation and migration and keratin expression.22,23 Fibroblasts establish such growth-promoting roles through paracrine crosstalk between NHOFs and NHOKs by cytokines such as heparin-binding EGF-like growth factor, interleukin-1α, and transforming growth factor-β1. 24 This function requires an optimal fibroblast density, as the presence of either excessive or inadequate numbers of fibroblast will adversely affect epithelium morphogenesis, leading to differentiation markers being inappropriately expressed. 25 In our models, the optimal fibroblast seeding density to support an anatomically representative epithelial layer was found to be 2 × 105 cells per model. Keratinocyte senescence may also impact the capacity to achieve anatomically representative epithelia in tissue culture models. Due to sustained telomerase expression within primary cells, sequential subculturing may restrict the capacity of keratinocytes to divide once seeded into a 3D model, and therefore it is recommended that the use of keratinocytes in tissue substitute constructs is limited to passage 3 or less. 26
The proper functionality of keratinocytes is important not only for epithelial layer formation but also for epithelial-connective tissue attachment and cell–cell adhesion, which are essential for achieving an accurate, functional mucosal substitute. Our data, consistent with data from other studies, have confirmed that cells within the 3D models actively synthesized the ultrastructural components, including desmosomes, hemidesmosomes, and basement membrane, required for this structural stability.27,28
Although COL1 is a nonspecific marker of osteogenesis, relatively high levels were expressed in our model compared to other osteogenic genes; this may be due to a number of reasons. First, this marker constitutes the most abundant component of bone extracellular matrix (90% of the organic component) and becomes upregulated during bone formation. 29 Second, ascorbic acid was added as a component of the culture medium to maintain the osteoblastic phenotype of bone-derived cells; ascorbic acid is known to stimulate of cell growth and collagen synthesis in osteoblasts. 30 The hydroxylation of proline residues of procollagen is increased to ∼40% by ascorbate, which is known to stabilize the collagen triple helix. 31 Third, although COL1 is the main organic component secreted by osteoblasts, it is also produced in abundance by fibroblasts, 32,33 and therefore the fibroblastic component of the model may have contributed to the collagen levels observed.
We undertook analysis of further osteoblastic markers, including OC, OP, ON and ALP. ALP, like COL1, is expressed in tissues other than bone and therefore its expression cannot be considered specific, although it is synthesized by osteoblasts and has been used to assess osteoblast phenotype and matrix mineralization. 34 Conversely, OC, OP, and ON are major noncollagenous, bone-specific proteins that play profound roles in bone formation. 29 ON, in particular, is localized to mineralized bone trabeculae and is present at higher concentrations within extracellular matrix than in osteocytes. It selectively binds to COL1 and the resultant ON-collagen complexes initiate mineral phase deposition by binding synthetic apatite crystals and free calcium ions. 35 Interestingly, ON can be demonstrated in active osteoblasts and osteoprogenitor cells, as well as in young osteocytes, but not in aged, quiescent osteocytes; the protein is therefore a reliable marker of functional osteoblasts. 36 The simultaneous expression of ON and its encoding gene, SPARC, strongly suggests that HOBs maintained their phenotypic characteristics despite a protracted culture period of 2 months.
The expression of all osteoblast-associated molecules varies over the different stages of bone development, and therefore the expression profiles observed in our models, which were cultured over a relatively short period compared to that of normal human bone turnover, may vary somewhat to that observed in vivo. For example, ALP increases in the initial stages of bone formation, yet decreases as mineralization progresses, whereas OP is first detected in young bone, while OC appears toward the end of the mineralization process. 37
Conclusion
This study demonstrates that tissue-engineered trilayered reconstructs based on primary HAOBs cultured in porous HA/TCP scaffolds adhered to collagen-embedded gingival fibroblasts, overlain with primary gingival keratinocytes cultured at the air/liquid interface, were able to mimic the native alveolar bone and overlaying full-thickness mucosal structures. The engineered combined hard and soft tissue provides scope to act as a valuable alternative to 2D and animal models for various in vitro and potential in vivo applications. Further experiments are underway to assess the suitability of this 3D model for in vitro evaluation of bone invasion of oral cancer as well as development of in vitro tissue-engineered models of osseointegrated dental implants using the 3D composite bone mucosal system.
Footnotes
Acknowledgments
The authors thank Abdurahman El-Awa, Yusuf Alzayani, Ei Leen Lim, Claire Field, and all the other surgeons who provided us with bone chips and oral mucosa tissue. They also thank Kirsty Franklin for her help and support in the tissue culture laboratory, Chris Hill for his assistance with electron microscopy services, and Rebecca Goodchild and Ceramisys, Ltd. (UK) for providing the bone scaffolds.
Disclosure Statement
No competing financial interests exist.