
Abstract
Tissue antigenicity represents the main limitation for the use of xenografts in clinical practice. To eliminate xenoantigens and avoid graft rejection in human, decellularization is often used to remove all immunoreactive components from the extracellular matrix (ECM). After decellularization, acellular scaffolds are required to be investigated regarding the presence of antigens, but commonly used detection methods solely focus on known xenoantigens such as alpha Gal (Galα1,3-Galβ1–4GlcNAc-R) or major histocompatibility complex-I (MHC-I). However, there are unknown xenoantigens that escape the standard methods. To evaluate the immunological potential of xenogenic tissues, new in vitro methods need to be developed. Therefore, we established a novel human serum-based approach, including dot blot, Western blot, immunohistochemistry, and enzyme-linked immunosorbent assay (ELISA). With these methods, we analyzed protein extracts and tissue samples of native and decellularized bovine carotid arteries. All methods verified an effective removal of potential immunogens from the ECM through decellularization, and relative quantification with ELISA showed that 99.9% (p < 0.01) of antigenic components were successfully eliminated. We compared our human serum-based methods with commonly used assays for the detection of alpha Gal and MHC-I. Our results showed highly increased sensitivity for xenoantigens using the human serum antibody pool. This novel in vitro detection system allows the direct determination of the immunogenic potential of xenografts and is a vast improvement in comparison to the methods used so far. That way, it is possible to optimize the decellularization process to prevent hyperacute graft rejection in patients.
Introduction
B
To reduce the immunogenic potential, xenogenic tissue has been subjected to several methods that remove or mask antigens while preserving structural integrity and functional properties. The most commonly used method is the chemical treatment with glutaraldehyde that crosslinks proteins and masks xenogenic antigens.3–5 Glutaraldehyde prevents hyperacute and acute xenograft rejection, but delayed immune recognition and graft degeneration occur.6–9
Decellularization focuses on the removal of cells from xenogenic tissues. The success of cell removal is dependent on the material, and therefore, a variety of decellularization protocols exist for different tissues,1,10 such as heart valves,11,12 pericardium,13,14 bovine 15 and equine carotid arteries,16,17 cartilage,18,19 and dermis.20,21 The concept of tissue decellularization to reduce the immunogenic potential of xenografts is based on the assumption that cell removal equals antigen removal. Since xenoantigens have been shown to remain in acellular scaffolds,22–24 the focus has shifted to decellularization techniques that include antigen removing steps.2,9,25–29
In particular, DNA, the alpha Gal epitope (Galα1,3-Galβ1–4GlcNAc-R), and major histocompatibility complex-I (MHC-I) are considered to be responsible for the persistent antigenicity of decellularized xenografts. Although the immunogenic potential of DNA is not verified, it is discussed controversially.30–33 The alpha Gal epitope and MHC-I, on the contrary, are known xenoantigens that trigger hyperacute and acute rejection mechanisms and ultimately cause graft degeneration and failure.22,30,33–35
Methods commonly used for analyzing decellularized xenografts regarding cell removal and immunogenic potential include histological assessment of residual nuclei such as hematoxylin and eosin staining,11,16,19,28,31,34,36–39 DAPI staining,31,34,37,39,40 quantification of DNA content,11,16,19,34,36,37,39,40 analysis of DNA fragmentation,31,34,37,39 and quantification of residual cellular proteins.16,41–43 However, these methods only detect cellular components with neither confirmed nor denied antigenicity. Methods that focus on a more thorough approach to determine the absence of known antigens such as alpha Gal and MHC-I include immunohistochemistry (IHC),22–24,28,29 enzyme-linked immunosorbent assay (ELISA),11,25,27,29,44,45 and immunoblotting.16,24,26,36,46,47
Unfortunately, detection of known xenoantigens such as alpha Gal and MHC-I gives no information about the persistence of unknown antigens. As previously reported, even organs derived from alpha Gal knockout pigs are subject to acute humoral xenograft rejection48–52 and elevated levels of non-Gal antibodies have been observed after implantation,49,50,53 indicating the presence of non-Gal xenoantigens. Methods for the identification of unknown antigens in decellularized tissues include complex immunoproteomic approaches such as two-dimensional gel electrophoresis and mass spectrometry.8,16,54,55 These methods are expensive and time-consuming, and the antigenicity of identified proteins still needs to be confirmed. Therefore, to investigate the actual immunogenic potential of decellularized tissues, in vivo tests are necessary.16,19,37,38,45,56
There is an unmet need for reliable and fast in vitro methods to investigate xenoantigenicity and to assess the entire immunogenic potential of decellularized grafts aside from known antigens. Consequently, we developed novel human serum-based methods and used a human serum pool, collected from ∼40 donors to minimize biological differences. The serum naturally contains a high-reactive, polyclonal antibody composition, which is associated with the adaptive immune response and responsible for hyperacute graft rejection. 57 We used that composition as a primary antibody in dot blot, Western blot, IHC, and ELISA and detected antibodies bound to xenoantigenic proteins with polyvalent, peroxidase-linked immunoglobulins. The results of these analyses were compared to common techniques such as alpha Gal and MHC-I staining.
Materials and Methods
Tissue preparation and decellularization
Bovine carotid arteries (BACs) were obtained from a local slaughterhouse. Perivascular tissue was carefully removed from the vessels on the same day. Branches of the BACs were ligated with 2-0 Mersilene (Johnson & Johnson Medical GmbH, Norderstedt, Germany). Full-length arteries were treated with 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) detergent in phosphate-buffered saline (PBS) with 1 M NaCl and 25 mM EDTA as described before. 15 Briefly, the vessels were integrated in a perfusion chamber and incubated with detergent at 37° for 16 h. Furthermore, we applied a two-step treatment for 16 and 22 h (16 + 22 h) with an additional rinsing step after 16 h. BACs were extensively rinsed with 0.9% NaCl and sterilized after decellularization. Samples were taken from every BAC and stored at −20°C until protein extraction or fixed over night in Carnoy buffer (ethanol, chloroform, acetic acid; 6:3:1) for IHC.
Isolation of bovine endothelial cells
Freshly prepared and ligated BACs were rinsed with PBS to remove blood residues. BACs were filled with collagenase solution (0.1% collagenase type II in PBS) and were incubated for 5 min at 37°C to remove endothelial cells. Cells were washed out with medium M199 (Biochrom GmbH, Berlin, Germany) and were collected in a reaction tube. For one BAC, the procedure was repeated three times. Each fraction was centrifuged at 400 g for 5 min, and the cell pellet was resuspended in medium M199 (10% FKS, 2%
Protein extraction and quantification
One hundred milligrams of BAC were homogenized in 500 μL TE-buffer (10 mM Tris–HCl, 1 mM EDTA, pH 7.2) with steal beads at 3000 rpm for 1 min (Microtube Homogenizer; GeneOn GmbH, Ludwigshafen, Germany), followed by 15 min of ultrasonification. The tissue was sedimented by centrifugation, and the supernatant was used to determine protein content. The content of soluble proteins was measured using the Bradford assay (Bio-Rad Laboratories GmbH, Munich, Germany).
Dot blot analysis
Protein extracts of native and decellularized BACs were used for dot blot analysis. Five microliters of extract were dropped on a nitrocellulose membrane and dried. Membranes were treated with blocking buffer (Table 1) for 30 min at room temperature. Primary antibodies were diluted in blocking buffer and incubated for 16 h at 4°C. After rinsing in PBS-T (PBS with 0.05% Tween-20), secondary antibodies were diluted and added for 1 h at room temperature. Rinsed membranes were incubated with the Pierce™ DAB Substrate Kit (Thermo Fisher Scientific, Inc., Waltham, MA) for ∼20 min.
BSA, bovine serum albumin; ELISA, enzyme-linked immunosorbent assay; IHC, immunohistochemistry; MHC-I, major histocompatibility complex-I; PBS-T, phosphate-buffered saline with 0.05% Tween-20.
Sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blot analysis
Protein extracts of native and decellularized BACs were used for sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. Protein samples were mixed with 6 × Laemmli buffer and heated at 95°C for 5 min. Ten microliters of each sample and 10 μL of Protein Ladder PS10 PLUS (GeneOn GmbH) were loaded into an SDS-gel (5% stacking gel, 10% separating gel). Electrophoresis was performed in running buffer (0.1 M Tris-Base, 0.1 M HEPES, 3.5 mM SDS) at room temperature for ∼45 min using a constant voltage (150 V). Gels were stained with Coomassie brilliant blue or placed in transfer buffer (25 mM Tris-base, 200 mM glycine, 10% methanol) for Western blot analysis.
For Western blot analysis, six layers of Whatman paper and a nitrocellulose membrane were equilibrated in transfer buffer. The blotting was performed at constant voltage (25 V) for 20 min. Membranes were washed in PBS-T for 5 min. Membranes were treated with blocking buffer (Table 1) for 60 min at room temperature. Primary antibodies were diluted in blocking buffer and incubated for 16 h at 4°C. After rinsing with PBS-T, secondary antibodies were added for 2 h at room temperature. Rinsed membranes were incubated with the Pierce DAB Substrate Kit for ∼20 min. The MHC-I antibody was not suitable for the detection of denatured proteins using Western blot analysis.
IHC analysis
Native and decellularized BAC samples were fixed for 16 h in Carnoy buffer (60% ethanol, 30% chloroform, 10% acetic acid), dehydrated using increasing ethanol concentrations, and embedded in paraffin. Sections of 5 μm were used for staining. Paraffin sections were dried at 62°C for 2 h. Afterwards, slides were incubated three times for 10 min in xylene and rehydrated in decreasing alcohol concentrations. After rinsing with distilled water, endogenous peroxidases were inactivated in 0.3% H2O2 in PBS for 10 min. Slides were washed in PBS-T for 5 min and were treated with blocking buffer at room temperature. After an additional washing step, slides were incubated at 4°C with a primary antibody solution and secondary antibodies were added for 1 h at room temperature. Exact specifications can be found in Table 1.
Slides were rinsed and incubated with the UltraVision Quanto HRP DAB Detection System (Thermo Fisher Scientific, Inc.) for ∼20 min. After rinsing with distilled water, cell nuclei were stained with hematoxylin for 3 min, which were blued during 10 min in warm water. Finally, sections were embedded in Roti-Mount Aqua (Carl Roth GmbH, Karlsruhe, Germany), dried, and documented by light microscopy (Axiovert 25; Carl Zeiss AG, Oberkochen, Germany).
Slides with endothelial cells were fixed for 1 h with 70% ethanol. IHC staining started with the decreasing alcohol series similar to paraffin sections.
ELISA
Protein extracts of native and decellularized BACs were used for ELISA. High-binding 96-well plates (Corning, Inc., New York, NY) were coated with 100 μL of protein samples for 1 h. Protein extracts from native BACs were diluted in 12 steps from 1:5 to 1:10,000. Protein samples of decellularized BACs were not diluted. All samples were analyzed in duplicate.
Wells were treated with 150 μL blocking buffer (Table 1) for 30 min at room temperature and washed in PBS-T three times for 5 min. One hundred microliters of primary antibody solution was incubated for 2 h at room temperature. After rinsing with PBS-T, 100 μL of secondary antibody solution were added for 1 h at room temperature. After rinsing, wells were incubated with 100 μL Ultra TMB Reagent (Thermo Fisher Scientific, Inc.) for ∼20 min. Finally, 100 μL stop reagent was added and the extinction was measured at 450/620 nm.
Statistic
All statistical data were analyzed with GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Outliers were identified using Grubb's test, and normal distribution was analyzed by D'Agostino-Pearson test. Differences between mean values were investigated for significance by t-test. The subsequent data are shown as mean ± standard deviation. A p-value of *p < 0.05 was considered to be statistically significant and **p < 0.01 to be very significant.
Results
In vitro analysis of alpha Gal-epitopes or MHC-I-molecules is often used to evaluate the immunogenicity of xenografts, but a lot of unknown proteins are able to activate the immune system. The aim of this study was to develop a detection system that allows the quantitative determination of xenograft's immunogenic potential in vitro without focusing on individual known xenoantigens.
Dot blot analysis
At first, the immunogenic potential of BAC proteins was analyzed with the dot blot technique. This analysis is suitable for rapid detection of proteins in their tertiary structure.
Serum, alpha Gal, and MHC-I staining showed a positive signal to protein extracts from native BACs, but it was more intense with serum. Furthermore, two protein extracts from decellularized BACs were compared. The alpha Gal and MHC-I dot blot showed no signal in decellularized samples, however, the human serum dot blot showed differences between the 16-h and 16 + 22-h decellularization process. The 16-h decellularized BAC showed a strong signal and controls were negative (Fig. 1).

Dot blot protein analysis of native and decellularized BACs. Proteins were extracted from nondecellularized (native), 16-h decellularized, and 16 + 22-h decellularized BACs. Five microliters of protein extracts or TE-Buffer (control) was applied onto a nitrocellulose membrane and stained with human serum, anti-MHC-I, or anti-alpha Gal antibodies. Secondary staining was performed with peroxidase-linked antibodies and detected with DAB substrate. BACs, bovine carotid arteries; MHC-I, major histocompatibility complex-I.
SDS-PAGE and Western blot analysis
BAC proteins were separated in size with SDS-PAGE and analyzed using Coomassie brilliant blue staining and human serum Western blot. Coomassie brilliant blue staining was used to visualize all proteins retained in decellularized BACs and Western blot analysis to detect potentially immunogenic proteins.
All BAC proteins with a size up to 200 kDa were detected with Coomassie. Many of these native BAC proteins were stained positive with human serum. After decellularization, proteins were strongly reduced in samples of both processes. In BAC samples treated for 16 h, Coomassie staining detected protein bands from 35 to 48 kDa and 75 to 135 kDa. Human serum Western blot revealed positive protein bands between 63 and 120 kDa. Samples of 16 + 22-h decellularized BACs showed no protein bands (Fig. 2).
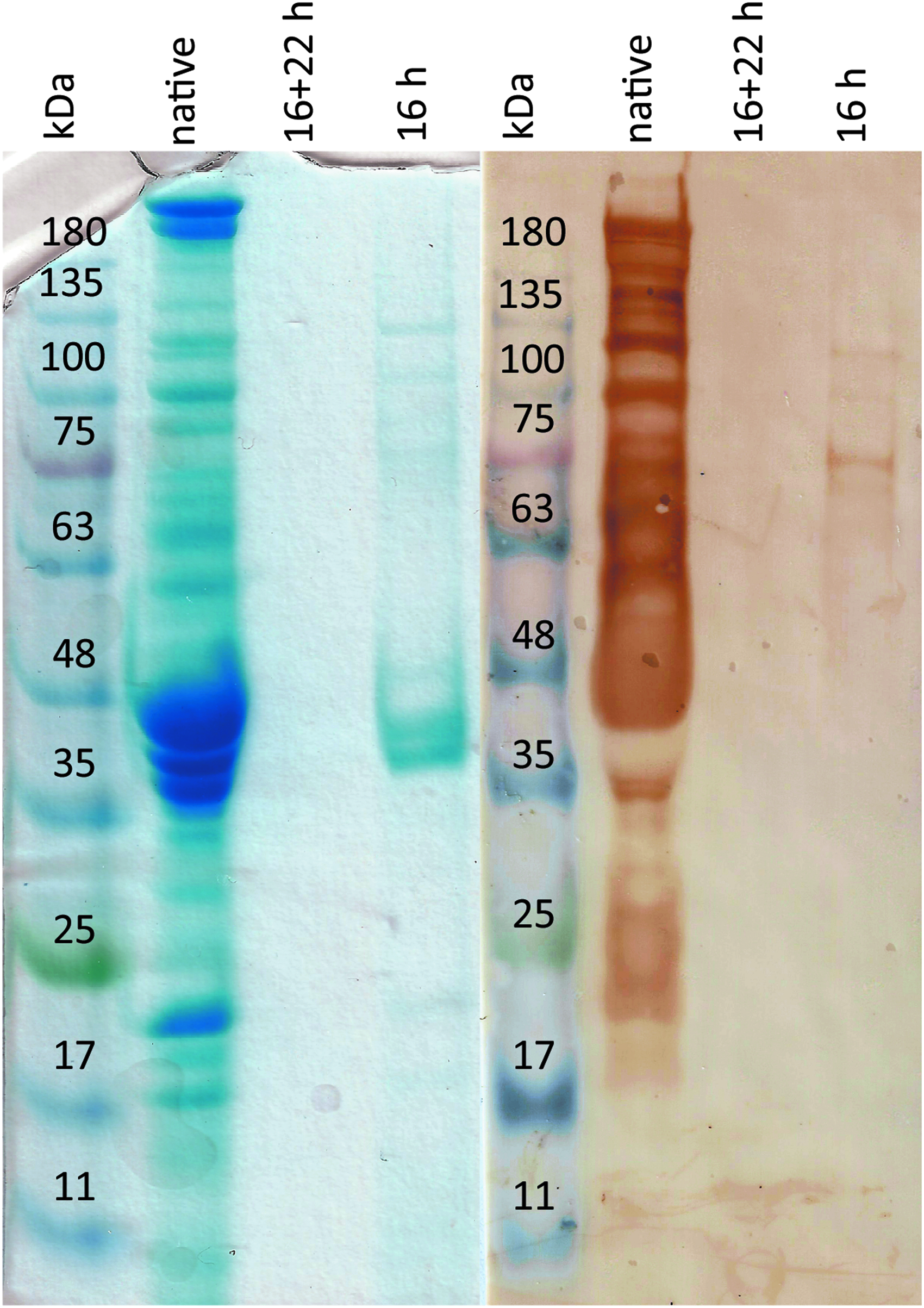
SDS-PAGE and Western blot protein analysis of native and decellularized BACs. Proteins were extracted from nondecellularized (native), 16-h decellularized, and 16 + 22-h decellularized BACs. Ten microliters of extracts or 10 μL protein ladder was loaded onto a 10% SDS-gel. The protein gel was colored with Coomassie brilliant blue or blotted onto a nitrocellulose membrane. The membrane was stained with human serum. Secondary staining was performed with peroxidase-linked antibodies and detected with DAB substrate. SDS-PAGE, sodium dodecyl sulfate/polyacrylamide gel electrophoresis.
Immunohistochemical analysis
Immunohistochemical staining of tissue sections was established to detect and localize immunogenic proteins.
Cell cultures of bovine endothelial cells were fixed with 30% ethanol and stained with human serum, anti-alpha Gal, and anti-MHC-I antibodies. Bovine endothelial cells were tested positive for alpha Gal and MHC-I, but staining with human serum was more intensive (Fig. 3).
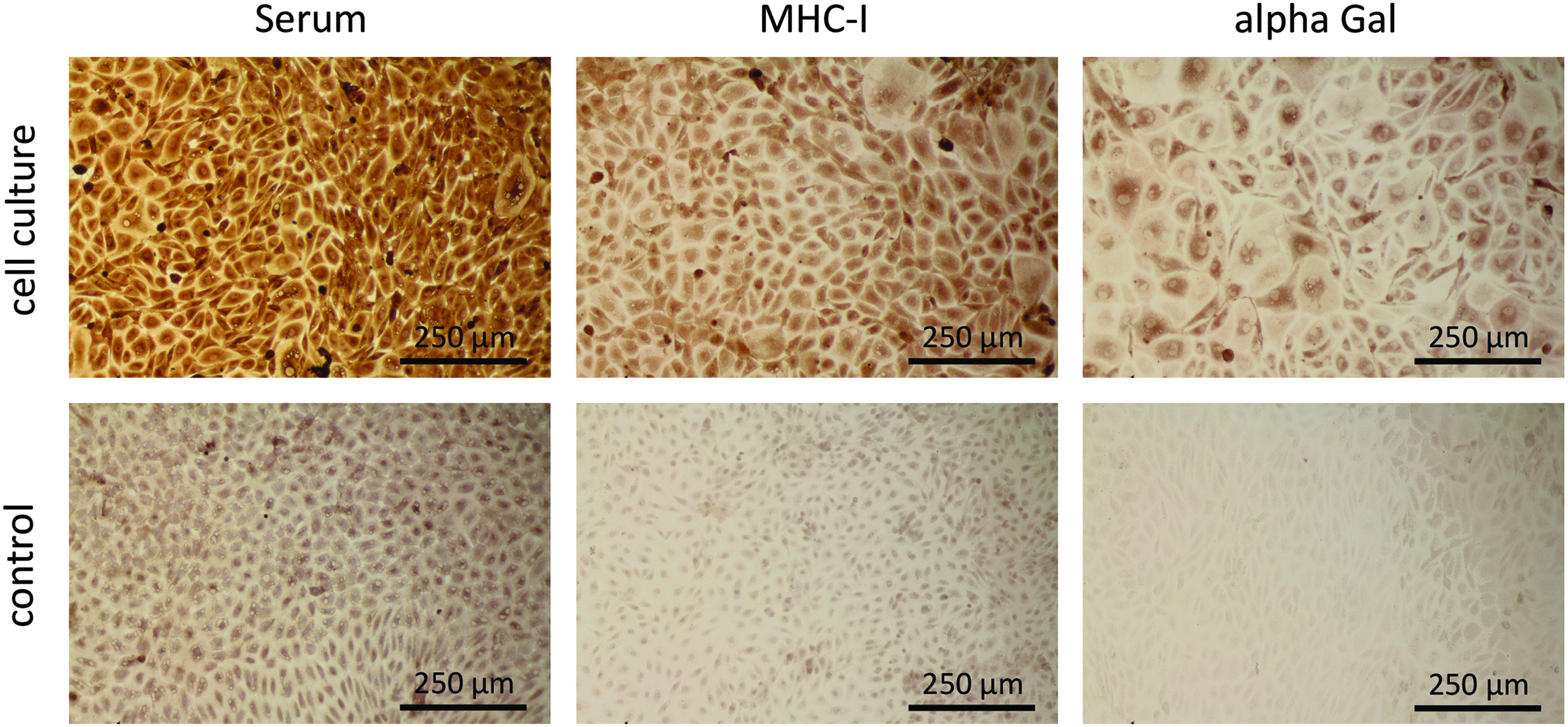
Immunohistochemical staining of bovine endothelial cell cultures with human serum, MHC-I, and alpha Gal. Cells were isolated from BACs and cultured on coverslips. Cells were fixed with ethanol and stained with human serum, anti-MHC-I, or anti-alpha Gal antibodies. Primary staining was not performed for controls. Secondary staining was done with peroxidase-linked antibodies and detected with DAB substrate.
Furthermore, analysis of whole tissue sections of native BACs with human serum resulted in a strong detection of immunogens on intimal endothelial cells and medial smooth muscle cells. MHC-I epitopes were identified on endothelial cells and smooth muscle cells but the signal was poor compared to human serum staining. Alpha Gal epitopes were only detected on endothelial cells. Decellularized BACs showed no alpha Gal epitopes and MHC-I molecules. However, analysis with human serum presented a very light staining of the intimal site (Fig. 4).

Immunohistochemical staining of BAC sections with human serum, MHC-I, and alpha Gal. BACs were analyzed before (native) and after 16 + 22-h decellularization. Tissue samples were fixed in Carnoy buffer, dehydrated, and embedded in paraffin. Five micrometer tissue sections were stained with human serum, anti-MHC-I, or anti-alpha Gal antibodies. Primary staining was not performed on controls. Secondary staining was done with peroxidase-linked antibodies and detected with DAB substrate. Arrows indicate a positive staining.
ELISA
The human serum ELISA was established for a semiquantitative analysis of potentially immunogenic proteins.
Ninety-six-well plates were coated with protein extracts of native BACs from 0.00037 to 3.7 mg/mL and human serum was used as primary antibody. The measuring range was identified between protein levels of 2 and 70 μg/mL. The linear range of the standard curve can be described as a natural logarithmic function: y = 0.443 ln(x) + 3.0849 (Fig. 5).

Standard curve of human serum ELISA. The immunogenic potential of native BACs was detected by semiquantitative ELISA. Plates were coated with diluted protein extracts of native BACs from 0.00037 to 3.7 mg/mL and blocked with 0.5% fish gelatin. After incubation with human serum, secondary staining was performed with peroxidase-linked antibodies and detected with TMB substrate. Absolute protein concentrations determined by Bradford assay were logarithmically plotted on x-axis, extinction of each sample on y-axis. Results are shown as mean ± SD (n = 3). ELISA, enzyme-linked immunosorbent assay; SD, standard deviation.
Human serum ELISA was used to compare the immunogenic potential of native and decellularized BACs. The immunogenic potential is given as percentage of native BACs (native BACs represent 100%). Decellularization with CHAPS for 16 or 16 + 22 h yielded in a very significant reduction of the immunogenic potential of 99.3% (16 h, p < 0.01) and 99.9% (16 + 22 h, p < 0.01). Extended decellularization of 16 + 22 h showed a significant reduction (p < 0.05) of immunogenicity compared to the 16-h process (Fig. 6).
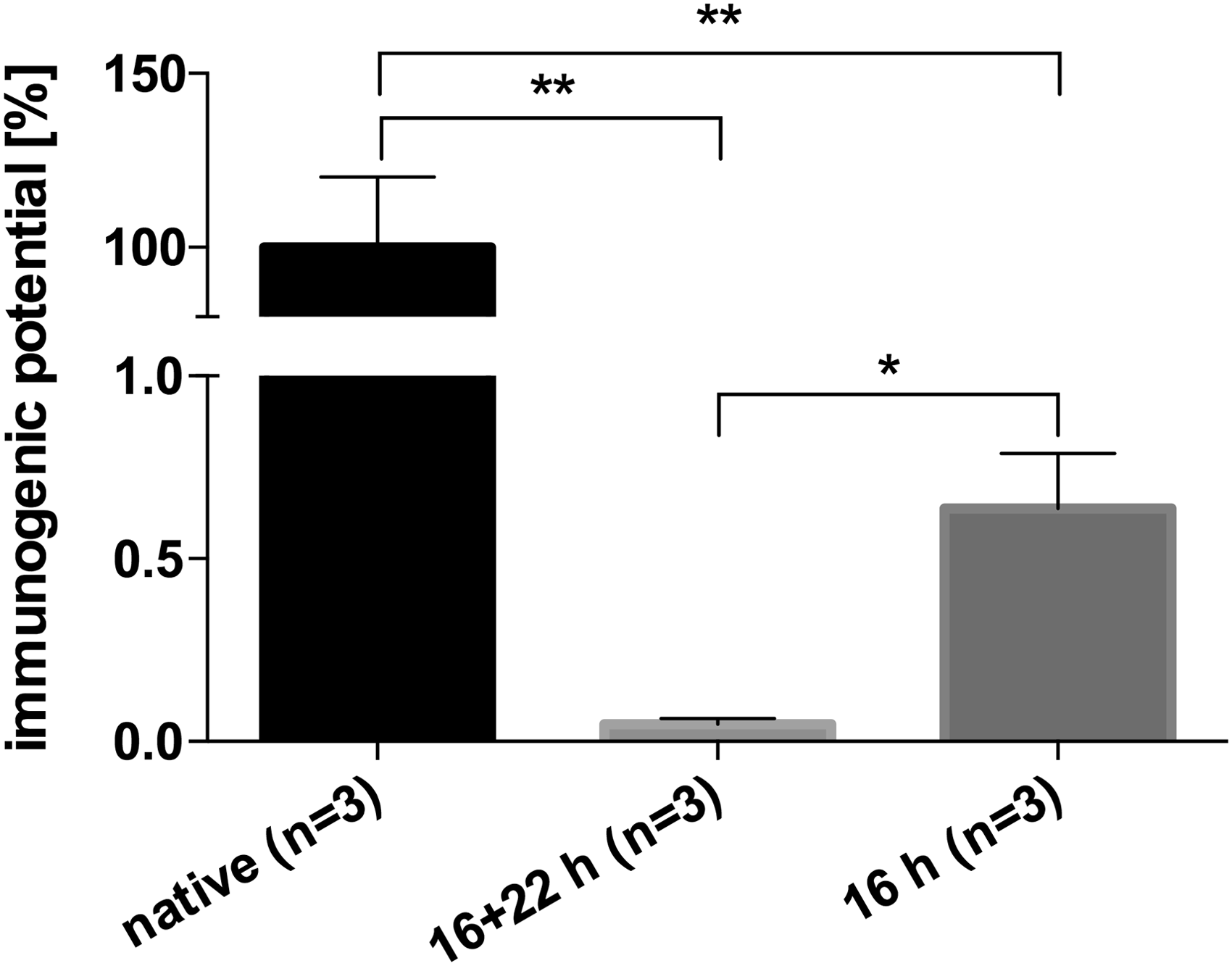
Analysis of BACs with human serum ELISA. The immunogenic potential of nondecellularized (native), 16 + 22-h decellularized, and 16-h decellularized BACs was detected by semiquantitative ELISA. Plates were coated with diluted protein extracts of native and decellularized BACs and blocked with 0.5% fish gelatin. After incubation with human serum, secondary staining was done with peroxidase-linked antibodies and detected with TMB substrate. Results are shown as mean ± SD, *p < 0.05 and **p < 0.01.
The same protein samples of native and decellularized BACs were analyzed with semiquantitative alpha Gal and MHC-I ELISA and both showed a 20–30 times lower extinction compared to the human serum ELISA. Alpha Gal and MHC-I were not detectable in decellularized samples.
Discussion
Due to chronic organ or tissue failure, transplantation is required. Unfortunately, homografts are poorly available and the main limitation for the use of xenografts is immunological rejection as a result of the phylogenetic differences between donor and recipient.9,17 The immunogenicity of animal tissues can be reduced using different methods, such as chemical treatment with glutaraldehyde or decellularization. The aim of decellularization is the complete removal of all cellular components from the extracellular matrix (ECM) to prevent activation of the innate and adaptive immune response.1,16 Evaluation of residual cellular components such as DNA via histological and quantitative methods is the most commonly used method to characterize an acellular tissue.11,16,19,28,31,34,36–39,41–43 We previously demonstrated the complete cell removal from decellularized BACs using DNA, RNA, and protein quantification. 15 However, conclusions concerning their immunogenicity are only indirectly possible, since antigenic components have been shown to remain in acellular scaffolds.22–24 Even detection of known antigenic determinants such as alpha Gal and MHC-I gives no information about the persistence of unknown xenoantigens and provides only a partial picture. Due to this reason, we developed an in vitro detection system that allows the quantitative determination of the immunogenic potential of xenografts aside from individual molecules with confirmed antigenicity.
To detect potential xenoantigens in vascular transplants, we used a human serum pool (from ∼40 donors) containing polyclonal IgM, IgG, and IgA antibodies, which are associated with primary immune response and hyperacute graft rejection, 57 as primary antibodies. We used a polyvalent antibody directed against human Ig(A, M, G) as secondary antibody and fish gelatin blocking buffer, since conventionally used proteins of mammalian origin cross react with antibodies from human serum (e.g., bovine serum albumin, dry milk). On this basis, we established human serum-based dot blot, Western blot, IHC, and ELISA to evaluate the immunogenic potential of native and decellularized BACs. Dot blot analysis was suitable for rapid detection of immunogenic proteins in their tertiary structure, whereas Western blot was established to characterize and identify potential immunogens depending on their size after denaturation. IHC staining was established to locate xenoantigens in tissue samples and to evaluate the ECM regarding its immunogenicity. ELISA was used as a semiquantitative method to detect antigenic determinants.
With these human serum-based methods, we compared the immunogenic potential of native and decellularized BACs. Native samples showed the strongest signal indicating the highest number of immunogens across all assays. Potential immunogens were effectively removed by intensified decellularization for 16 + 22 h, whereas decellularization for 16 h was incomplete and the remaining immunogens were successfully detected with human serum dot blot, Western blot, and ELISA. With the ELISA technique, we showed that 99.3% (16 h, p < 0.01) and 99.9% (16 + 22 h, p < 0.01) of antigenic components were effectively eliminated through decellularization. Extended decellularization of 16 + 22 h showed a significant reduction (p < 0.05) of immunogenicity compared to the 16-h process. The comparison of ELISA and dot blot analysis demonstrated that even the blotting technique can sensitively present <1% of remaining immunogens.
Furthermore, the results were compared to alpha Gal and MHC-I detection techniques. These molecules are common markers for immunogenicity, since transplant rejection is primarily triggered by antibodies against these xenoantigens.58–60 Using dot blot, IHC, and ELISA, alpha Gal and MHC-I were successfully identified in native BACs but not in decellularized tissue samples. Samples of the 16-h and 16 + 22-h process showed no differences. In contrast to that, human serum techniques detected known and unknown xenoantigens revealing considerably stronger signals in native BACs and differences between the decellularization procedures. This increased sensitivity is due to the human polyclonal antibody pool that sensitively detects all, even the known and unknown xenoantigens. Using ELISA, immunogenicity can be identified at protein concentrations up to 2 μg/mL. Potential xenoantigens were not detectable in 16 + 22-h decellularized BACs, including alpha Gal and MHC-I. The decellularization of BACs successfully removed potential immunogens from the ECM and therefore cellular residues as described before. 15
Earlier in vitro investigations were not sufficient to predict the immunogenic potential of xenografts in recipient, because only single antigenic molecules were considered. Previously reported studies from alpha Gal knockout pigs demonstrated that inactivation of alpha Gal does not prevent xenograft rejection, which indicates the presence of non-Gal xenoantigens.48–52 Human serum can be used to determine the immunogenic potential of xenografts leading to hyperacute rejection, which can only occur in vascularized grafts in the first 24 h after transplantation and is triggered by the presence of antidonor antibodies existing in the recipient before transplantation.59,61 In particular, high amounts of anti-alpha Gal IgM and IgA antibodies circulate in human serum and it is suspected that they are responsible for hyperacute rejection of nonprimate xenografts.16,35,44 In addition, human serum IgG antibodies can react with donor MHC-I, which may as well result in hyperacute vascular rejection and graft failure.59,62–64 With our human serum-based methods, it is possible to compare and evaluate various xenogenic proteins and tissues of different origin for their immunocompatibility. Thus, it is possible to predict hyperactue graft rejection at an early development stage, which may reduce testing of the prostheses in animal experiments.
However, there are some limitations to this approach. First of all, the human serum-based method cannot rule out acute and chronic rejection due to innate and cellular immune responses. Acute and chronic rejection occurs during weeks after transplantation when antidonor antibodies are produced.59,61,65,66 These examinations are only possible in animal models, for example, Helder et al. analyzed the clinical relevance of non-alpha Gal antigens in cross-species acute immune response by measuring antidonor antibodies in recipient 45 and Böer et al. immunized mice with soluble matrix components and determined the formation of specific serum antibodies. 16 Moreover, it is unclear whether cellular components are solely responsible for the recipients' immune response. The three-dimensional ECM has not been fully characterized, but biomolecules such as collagen, elastin, laminin, fibronectin, and hyaluronic acid have been successfully isolated and identified. 60 In this study, we were not able to show any immunogenicity of the ECM using IHC staining. Especially collagens and laminins are evolutionarily very ancient and highly conserved. 67 There are a number of articles relating to the immunogenicity of xenogenic collagen68,69 and elastin,69,70 but after years of using biological materials in humans there is no definite evidence. 60
It should be kept in mind that xenotransplantation is a promising field and often the only alternative for patients with tissue or organ failure although there is no guaranteed immunocompatibility. However, even an imperfect homograft can induce a strong cellular and humoral recipient's immune response. 71
Conclusion
Due to chronic organ or tissue failure, transplantation is required, but the main limitation to the use of xenografts is immunological rejection. To avoid activation of the innate and adaptive immune responses, decellularization is used to remove all cellular components from the ECM. Evaluation of acellular scaffolds' immunogenicity is often evaluated indirectly via DNA quantification. In this study, we developed an in vitro detection system that allows direct determination of the immunogenic potential of xenografts. We established human serum dot blot, Western blot, IHC, and ELISA and showed considerably stronger signals compared to common methods for the detection of immunogens. This increased sensitivity is due to the detection of known and unknown xenoantigens. Each of these human serum-based methods can be used to predict the immunogenic potential of xenografts leading to hyperacute rejection in recipient.
Footnotes
Acknowledgments
The excellent technical assistance of Hatice Kilinc, BSc, Jacqueline Sonnenberger, BSc, My Linh Nguyen, BSc, and Mrs. Elena Lochau is gratefully acknowledged. This work was supported by the BMWi (Bundesministerium für Wirtschaft und Energie) project KF3002402CS4.
Disclosure Statement
No competing financial interests exist.