
Abstract
Decellularized peripheral nerve has been proven to be an effective clinical intervention for peripheral nerve repair and a preclinical cell carrier after spinal cord injury. However, there are currently a lack of decellularization methods for peripheral nerve that remove cells and maintain matrix similar to the previously established, clinically translated technique (the Hudson method) that relies on the discontinued Triton X-200 detergent. Therefore, the aim of this study was to optimize a novel chemical decellularization method for peripheral nerves based on the currently available anionic detergent sodium deoxycholate. Sprague Dawley rat sciatic nerves were isolated, frozen in buffered solution, and then subject to sequential washes in water, salt buffer, zwitterionic detergents sulfobetaines -10 and -16, and varying concentrations of sodium deoxycholate (SDC). To optimize DNA removal after SDC decellularization, nerves were subjected to deoxyribonuclease (DNase) incubation and salt buffer washes. Immunohistochemical results demonstrated that utilization of 3% SDC in the decellularization process preserved extracellular matrix (ECM) components and structure while facilitating significantly better removal of Schwann cells, axons, and myelin compared with the Hudson method. The addition of a 3-h DNase incubation to the 3% SDC decellularization process significantly removed cellular debris compared with the Hudson method. Proteomic analysis demonstrated that our novel decellularization method based on 3% SDC +3-h DNase used in conjunction with zwitterionic detergents, and salt buffers (new decellularization method using 3% SDC + 3-h DNase, zwitterionic detergents, and salt buffers [SDD method]) produced a similar proteomic profile compared with the Hudson method and had significantly fewer counts of cellular proteins. Finally, cytotoxicity analysis demonstrated that the SDD decellularized scaffolds do not contain significant cytotoxic residuals as eluted media supported metabolically active Schwann cells in vitro. Overall, this study demonstrates that SDD decellularization represents a novel alternative utilizing currently commercially available chemical reagents.
Impact Statement
Decellularized nerves are clinically relevant materials that can be used for a variety of regenerative applications such as peripheral nerve and spinal cord injury repair. However, discontinuation of key detergents used in a proven chemical decellularization process necessitates the optimization of an equivalent or better method. This research presents the field with a novel chemical decellularization method to replace the previous validated standard. Scaffolds generated from this method provide an extracellular matrix-rich material that can be used in a variety of in vitro applications to understand cellular behavior and in vivo applications to facilitate regeneration after neural injury.
Introduction
Tissue-specific extracellular matrix (ECM)-based scaffolds are valuable tools for tissue engineering and regenerative medicine. ECM-based scaffolds are bioactive and biodegradable, and can retain mechanical properties on the same order of magnitude as the native tissue.1,2 The bioactivity and mechanical environment of ECM-based scaffolds provide native cues that have been shown to encourage cell adhesion,3,4 migration,3,5 proliferation,6,7 differentiation,4,6 and proregenerative responses such as angiogenesis.8,9 In the decellularization process, cells and immunogenic cellular components are stripped from the supporting proteinaceous ECM.1,2 The decellularization process leaves an immunologically tolerated scaffold that preserves the native structure and composition of the ECM of the native tissue.1,2 Generated scaffolds can then be used as interventions after injury,10,11 as in vitro cell culture platforms or test beds,12,13 or combined with other materials to make composite materials.14,15
Chemical decellularization is one of the most common means of decellularization.1,2 The amphipathic molecules present in detergents aggregate in solution to form micelles that can readily remove cellular contents by disrupting lipid–lipid, lipid–protein, and protein–protein interactions.1,16 One such detergent is Triton X-200, an anionic detergent, that was previously utilized as part of a detergent-based decellularization process for peripheral nerve (the Hudson method). 17 This optimized method utilizes repeated washes in buffered solutions, anionic detergent Triton X-200, and zwitterionic detergents sulfobetaine-10 (SB-10) and sulfobetaine-16 (SB-16). 17
Overall, the Hudson method effectively and reliably removed cellular components, such as myelin. 17 However, what differentiates this method from other nerve decellularization methods is the superior preservation of ECM components and architecture. 17 In particular, the Hudson method better preserves laminin and its associated basal lamina architecture, tube-like structures that run the length of peripheral nerve.17–19 This basal lamina microarchitecture has been implicated in facilitating peripheral nerve regeneration after injury17–19 and is therefore a critical component to maintain during decellularization.
Human decellularized peripheral nerve grafts generated using the Hudson method have been commercially available for peripheral nerve repair since 2007. 20 Such human decellularized nerve grafts are clinically effective with patients showing meaningful recovery rates of 75–100%.21–23 For example, a multicenter study reported by Brooks et al. implemented decellularized human nerve grafts in 132 patients with nerve defects ranging from 5 to 50 mm. 23 For all surgeries, motor and sensory nerves had 85.7% and 88.7% meaningful recovery, respectively. 23 Even in the longest defects (30–50 mm), the average recovery was 90.9% after an average follow-up time of 264 days. 23
More recently, decellularized peripheral nerve scaffolds have been utilized in preclinical spinal cord injury (SCI) models to help facilitate regeneration and functional recovery. When implemented as an ECM-based scaffold alone, hydrogels derived from decellularized peripheral nerves promote a more regenerative M2 macrophage response after contusion SCI in rodents. 11
Also after rodent SCI models, these scaffolds have been shown to be supportive therapeutic vehicles for different cell populations, including placental mesenchymal stem cells 24 and Schwann cells, 10 and neurotrophic factors such as brain-derived neurotrophic factor. 25 As a therapeutic vehicle, decellularized peripheral nerve scaffolds have supported transplanted cell survival,10,24 improved regeneration,10,24,25 and improved functional recovery. 24 Taken together, this material is promising for future neural regeneration research both as an intervention and also as an in vitro test bed to better model peripheral nerve tissue.
Unfortunately, Triton X-200, the key anionic detergent within the Hudson method is no longer commercially available from its only manufacturer and relevant volumes would not be feasibly synthesized locally. A suitable replacement for Triton X-200 within this decellularization process has not been identified. Currently, reported alterations in the Hudson method include substituting Triton X-10026 and omitting Triton X-200 from this process. 27 In both cases, the decellularization efficacy was compromised leading to poorer cellular removal compared with the original Hudson method. Discontinuation of Triton X-200 highlights the need to identify appropriate replacement detergents and conduct a broad assessment of decellularization efficacy to develop a peripheral nerve decellularization method that facilitates at least equivalent cellular removal and ECM preservation compared with the validated Hudson method.
Sodium deoxycholate (SDC) is an alternative anionic detergent commonly used in chemical decellularization and has been successful in decellularizing multiple tissue types. 2 The objective of this study was to optimize a novel chemical decellularization method for peripheral nerve that is based on the Hudson method, but implements commercially available reagents (SDC). This method should be at least equivalent in terms of cellular removal and preservation of key ECM components and architecture, such as the basal lamina.
Overall, replacing Triton X-200 with 3% SDC and a 3-h incubation in deoxyribonuclease (DNase) (new decellularization method using 3% SDC + 3-h DNase, zwitterionic detergents, and salt buffers [SDD method]) results in approximately equivalent decellularization profiles. This finding is supported by immunohistochemical analysis, label-free quantitative proteomics, Western blotting, and cytotoxicity testing. The SDD provides the field with a novel peripheral nerve decellularization method that matches or surpasses the previous validated method of decellularization.
Materials and Methods
Tissue procurement and preparation
All animal work was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Florida. Male Sprague Dawley rats (250–300 g) were obtained from Charles River (Wilmington, MA) and cared for by the Animal Care Services in accordance with IACUC standards and the Animal Welfare Act. Rats had access to 12-h light/dark cycles and standard water and food. Before nerve harvest, rats were euthanized using the American Veterinary Medical Association guidelines using carbon dioxide. Sciatic nerves were harvested aseptically, and epineurium sheaths were removed from the nerves using sterile forceps. Nerves were then frozen in 1 × phosphate-buffered saline (PBS) at −20°C until decellularization.
Tissue decellularization
Decellularization of sciatic nerves was adapted from methods previously established.11,17 Briefly, nerves were subjected to washes in water, salt buffers, zwitterionic detergents SB-10 (Millipore Sigma) and SB-16, chondroitinase ABC (ChABC; Millipore Sigma), and an anionic detergent. The anionic detergent varied based on method. For the Hudson method decellularized nerves, 0.14% Triton X-200 (Dow Chemicals) was used. For optimization of a new detergent concentration, a high (5% w/v), medium (3% w/v), or low (1% w/v) concentration of SDC (Millipore Sigma) was used. Addition of DNase (Millipore Sigma) incubation to the SDC decellularization process consisted of adding 3-, 12-, or 24-h incubation in DNase and subsequent washes in salt buffer. All steps except enzyme (DNase and ChABC) incubations were conducted under orbital agitation (Table 1). More details concerning each step in this decellularization process, such as volume, concentration, wash duration, and other conditions, can be found in Table 1.
Summary of Decellularization Methods Utilized in This Study
Not included in Hudson method and initial SDC investigation.
RT, room temperature; PBS, phosphate-buffered saline; SDC, sodium deoxycholate; SB-10, sulfobetaine-10; SB-16, sulfobetaine-16; DNase, deoxyribonuclease; Y, yes; N, no.
Immunohistochemistry and semiquantitative analysis
For immunohistochemistry (IHC), after decellularization, nerves were fixed using 4% paraformaldehyde and stored in 30% sucrose for 1 week. Nerves were then trimmed, soaked in optimal cutting temperature (OCT) compound at 4°C overnight, and flash frozen at −80°C for at least 5 min. OCT blocks were then stored at −20°C until sectioning. Frozen sections were obtained using a Leica CM1950 cryostat (Leica Biosystems, Germany) with a tissue thickness of 10 μm. Sections were stained using antibodies against collagen-I (Millipore Sigma; C2456, 1:500), collagen-IV (Abcam; ab6586, 1:500), laminin (Millipore Sigma; L9393, 1:500), β-III tubulin for axons (Abcam; ab7751, 1:1000), S-100 for Schwann cells (Millipore Sigma; S2644, 1:500), myelin basic protein (MBP) for myelin (Millipore Sigma; M3821, 1:150), and 4′,6-diamidino-2-phenylindole (DAPI) for nuclei (Thermo Fisher Scientific; D1306, 1:1000). Negative controls (no primary antibody) were prepared to ensure autofluorescence or nonspecific staining was minimal.
A Zeiss Imager Z.2 fluorescence microscope (Carl Zeiss, Germany) was used to visualize staining and capture 20 × magnification images. Zen 2012 Image Processing software was used to semiquantitatively analyze the removal or preservation of components listed above by determining the average signal intensity over a defined region of interest (≥58,000 μm2) across the area of the nerve cross section. A minimum of three distinct nerves, four images per nerve, and one field of view were used for each stain. A percent difference in staining intensity compared with the Hudson method decellularized nerves was calculated for each protein probed and compared between groups.
dsDNA quantification
Decellularized nerves (n ≥ 3) were washed in water and lyophilized for 72 h. Samples were then weighed and DNA isolated using a DNeasy Blood and Tissue Kit (Qiagen, Germany) according to the manufacturer's instruction. Purified DNA content was then determined by the QuantiFluor dsDNA System (Promega) according to the manufacturer's directions. Fluorescence was read using a Biotek Synergy HT Microplate Reader (Biotek). Fluorescence values for each sample were compared to a lambda DNA standard curve to determine dsDNA content.
Cytotoxicity assessment
Cell culture media were eluted from nerves decellularized using the Hudson and SDD methods to measure cytotoxic side effects of residual detergents. For this assay, decellularized nerves (n = 3) were incubated in 200 μL Dulbecco's modified Eagle's medium/F12 (Thermo Fisher Scientific) +1% penicillin–streptomycin–amphotericin (Thermo Fisher Scientific) at 37°C for 72 h. The eluted medium was supplemented with fetal bovine serum (10%; Thermo Fisher Scientific), forskolin (0.1%; Millipore Sigma), bovine pituitary extract (0.14%; Thermo Fisher Scientific), and fibroblast growth factor (0.2%) (Thermo Fisher Scientific) and added to Schwann cells seeded in a 96-well plate. Cell metabolic activity was then assessed using an alamarBlue assay (Thermo Fisher Scientific) as per the manufacturer's instructions after 2 days in culture.
Further cytotoxicity testing was conducted to ensure cytocompatibility of the scaffold contents as described previously by Crapo et al. 28 Briefly, decellularized nerves (n = 3 Hudson method and n = 3 SDD method) were lyophilized. Nerves were then minced with microscissors and digested at a concentration of 1 mg dry weight tissues per milliliter of 1 mg/mL pepsin (Millipore Sigma) in 0.01 M hydrochloric acid (Millipore Sigma). Digests were neutralized using 1 M sterile sodium hydroxide (Millipore Sigma) and 10 × PBS. Decellularized homogenates were mixed with cell media to final protein concentrations of 100 μg/mL. These solutions were incubated with cultured Schwann cells for 24 h before metabolic activity was assessed by alamarBlue assay (Thermo Fisher Scientific).
Finally, the cytotoxic threshold of SDC solution on Schwann cells was determined by preparing eight concentrations of SDC ranging from 0% to 1.5% SDC in cell media. Schwann cells were cultured for 24 h in their respective concentration group, and alamarBlue was used to quantify metabolic potential. If normalized metabolic activity was <70%, the solution was deemed cytotoxic.
Proteomic analysis
Nerves processed using the Hudson and SDD methods were washed in water and then lyophilized for 72 h. Samples (n = 3) were sent to the University of Florida Mass Spectrometry Research and Education center for label-free quantitative proteomic analysis. Tandem mass spectra (mass spectrometry/mass spectrometry [MS/MS]) were analyzed using Sequest (XCorr only) (Thermo Fisher Scientific). Sequest was set up to search SwissProt_2017_06.fasta (467,231 entries) assuming the digestion enzyme trypsin. Sequest was searched with a fragment ion mass tolerance of 0.020 Da and a parent ion tolerance of 10.0 ppm.
Scaffold (version Scaffold_4.8.4; Proteome Software, Inc.) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability by the Peptide Prophet algorithm with scaffold delta-mass correction. 29 Protein identifications were accepted if they could be established at >99.0% probability and contained at least one identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm. 30 Proteins were annotated with Gene Ontology terms from the National Center for Biotechnology Information. 31
Western blotting
The Hudson method decellularized nerve (n = 3) and SDD method decellularized nerve (n = 3) were solubilized in 1 × RIPA lysis buffer (Millipore Sigma), protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific), and phenylmethylsulfonyl fluoride (Millipore Sigma). Solubilization was assisted by manual mincing, probe sonication (Cole-Parmer), and gentle agitation at 4°C. Western blots were conducted using the Wes automated capillary-based Western blotting system (Protein Simple). For these experiments, Protein Simple's 12–230 kDa separation module, 66–440 kDa separation module, anti-rabbit detection module, and anti-mouse detection modules were used. Samples were prepared according to the manufacturer's instructions.
To probe for key ECM proteins, antibodies for collagen-I (Millipore Sigma; C2456), collagen-IV (Abcam; ab6586), and laminin (Millipore Sigma; L9393, 1:100) were used. Collagen-I and collagen-IV were detected on samples run on the 66–440 kDa module, and laminin was detected on the 12–230 kDa module. Protein peaks were optimized before running samples in duplicate for each specific protein. Results were quantified and analyzed using Compass for SW software (version 4.0.1) (Protein Simple). For analysis, Compass software measures the area under the curve of electropherograms using standard peak fitting.
Statistics
For all experiments, at least three separate decellularized nerves were analyzed. For all experiments except proteomic and Western blot analysis, groups were compared using one-way analysis of variance and subject to Tukey's post hoc analysis using STATISTICA software (Dell, Inc.). All data presented depict the mean ± standard error. Differences were considered significant if p < 0.05. Comparison of proteomic analysis between nerves decellularized using the Hudson method and SDD method was conducted within scaffold (Proteome Software, Inc.) using Fisher's exact t-test, and significance was defined as p < 0.05. For Western blot analysis, STATISTICA software (Dell, Inc.) was used to administer an independent Student's t-test. Significance was defined as p < 0.05.
Results
Effect of increasing SDC concentration on cellular removal
Schwann cell, axon, myelin, and DNA removal during detergent-based processing (no DNase) were determined using semiquantitative analysis of IHC. DAPI staining revealed that nerves decellularized using 3% and 5% SDC processes had significantly greater nuclear content compared with nerves processed using the Hudson method (p < 0.05) (Fig. 1a, b). Staining was not characteristic of discrete nuclei but was diffuse throughout the scaffolds. Increasing the concentration of SDC did not improve DNA removal.

Nerves decellularized using the 1% SDC process contained significantly higher amounts of axonal staining compared with the Hudson method (p < 0.05) (Fig. 1e, f) while Schwann cell (S100) and myelin (MBP) staining were comparable (Fig. 1c, g). Increasing the concentration to 3% and 5% SDC resulted in significantly improved removal of Schwann cells, axons, and myelin compared with nerves decellularized with the Hudson method and 1% SDC process (p < 0.05) (Fig. 1c–h). Increasing the concentration of SDC from 3% to 5% did not further improve cellular removal (Fig. 1c–h).
Overall, implementing 3% SDC as a replacement for Triton X-200 within the Hudson method serves as a sufficient concentration to facilitate cellular debris removal comparable with or better than the traditional Hudson method, but remains insufficient at removing remnant DNA.
Effect of increasing concentration of SDC on ECM preservation
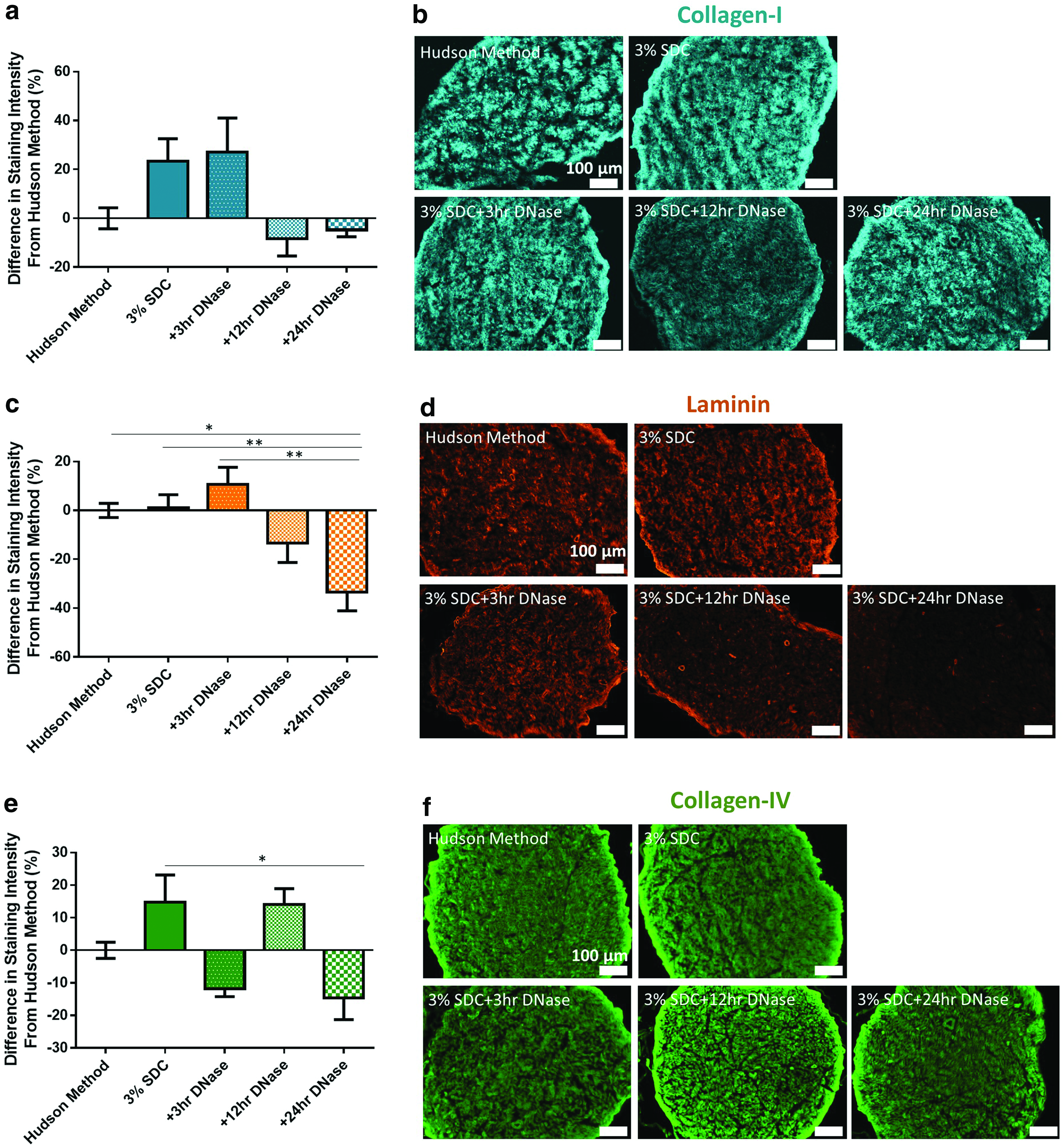
ECM preservation after decellularization was analyzed through semiquantitative analysis of collagen-I, collagen-IV, and laminin staining. Analysis demonstrates that collagen-I, collagen-IV, and laminin were not significantly removed by any concentration of SDC compared with the Hudson method. Collagen-IV (Fig. 2e, f) in nerves decellularized using the 5% SDC process had significantly brighter staining than those processed with the Hudson method (p < 0.05). This increase in signal is not thought to be due to differences in autofluorescence as control staining revealed a very low-level and consistent background in all groups (data not shown). Qualitatively, 5% SDC caused a greater structural damage compared with both 1% SDC and 3% SDC (Fig. 2b, d, and f) as noted by a disruption in ultrastructure and matrix continuity.

Overall, any concentration of SDC processing did not significantly remove key ECM components compared with the Hudson method. Collectively, we selected 3% SDC as a suitable replacement for Triton X-200 within the Hudson method. Although 3% SDC facilitates at least comparable cell removal and ECM preservation, DNA removal remains insufficient. To address this specific issue, an additional incubation in DNase was added to the 3% SDC decellularization process.
Effect of adding increasing DNase incubations on DNA and cellular removal
DNA removal was assessed by DAPI staining analysis and a quantitative dsDNA assay. DAPI staining (Fig. 3a, b) demonstrated that the addition of any duration of DNase incubation following the 3% SDC decellularization process facilitated significantly greater removal of DNA compared with nerves decellularized using the Hudson method (p < 0.05) and 3% SDC (no DNase) process (p < 0.05). Increasing the duration of DNase incubation did not cause further removal of DNA (Fig. 3a, b).
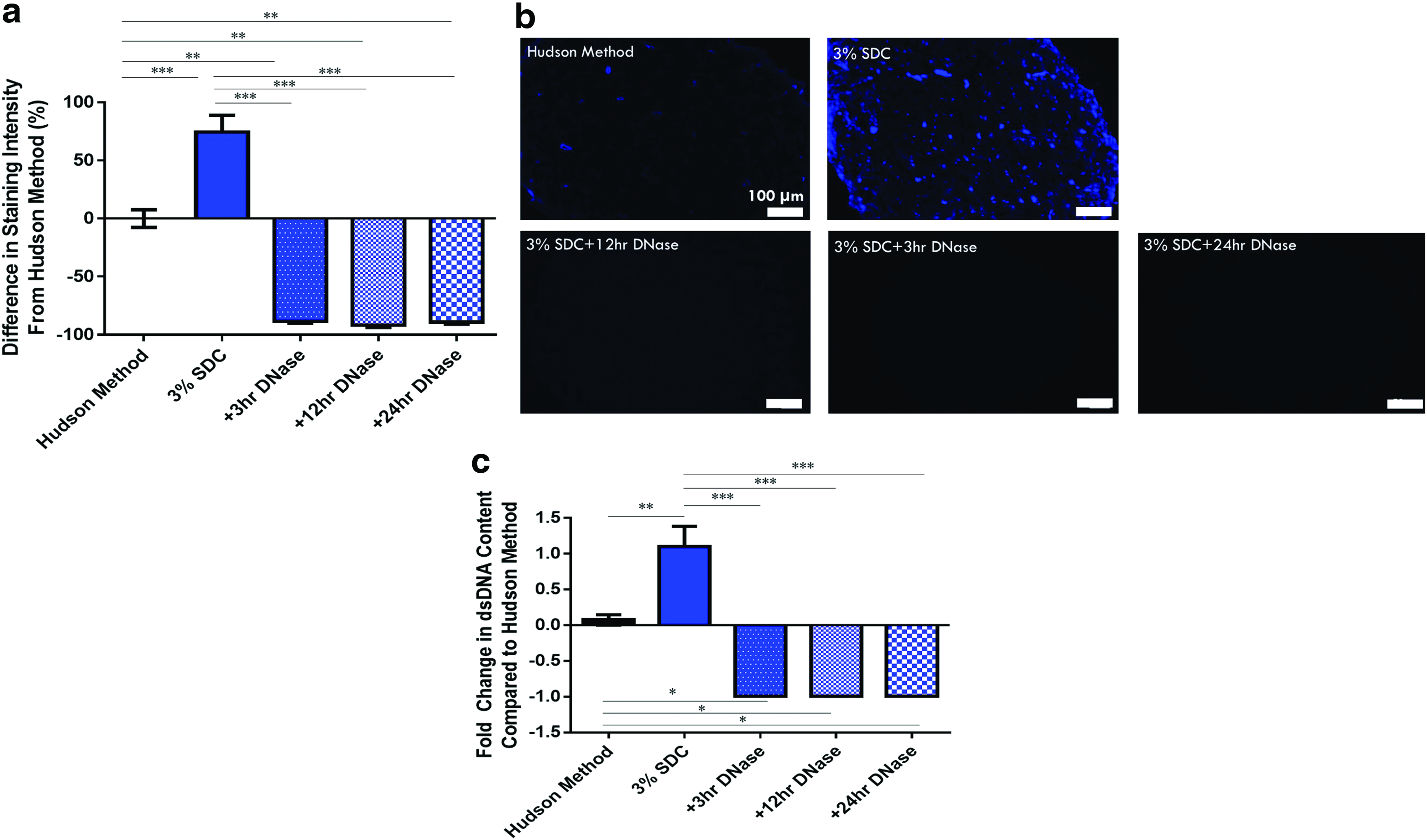
A similar trend was also seen with the quantitative results from the dsDNA assay (Fig. 3c). The addition of 3–24-h DNase incubations resulted in dsDNA levels compatible with current decellularization guidelines of 50 ng dsDNA/mg ECM proposed by Crapo et al. 1 Nerves decellularized with the 3% SD and DNase processes contained 13.65 ± 8.21 ng dsDNA/mg ECM, 16.82 ± 12.13 ng dsDNA/mg ECM, and 15.46 ± 5.45 ng dsDNA/mg ECM for 3-, 12-, and 24-h incubations, respectively.
Removal of other cellular components after the addition of DNase incubations was also studied to determine if further cellular removal occurred. All 3% SDC + DNase processes removed significantly more Schwann cells, axons, and myelin compared with the Hudson method (p < 0.05) (Fig. 4a–f). However, this effective removal in the 3% SDC + DNase processing groups was not from the addition of DNase incubations, as removal in all cases was comparable with nerves decellularized using the 3% SDC (no DNase) process.
Overall, a 3-h DNase incubation is sufficient to facilitate better DNA removal compared with both the 3% SDC (no DNase) decellularization process and the Hudson method. In addition, the addition of any duration of DNase incubation did not further improve removal of other cellular markers compared with the 3% SDC (no DNase) decellularization process, but still more effective than the Hudson method.
Effect of adding increasing DNase incubations on ECM preservation
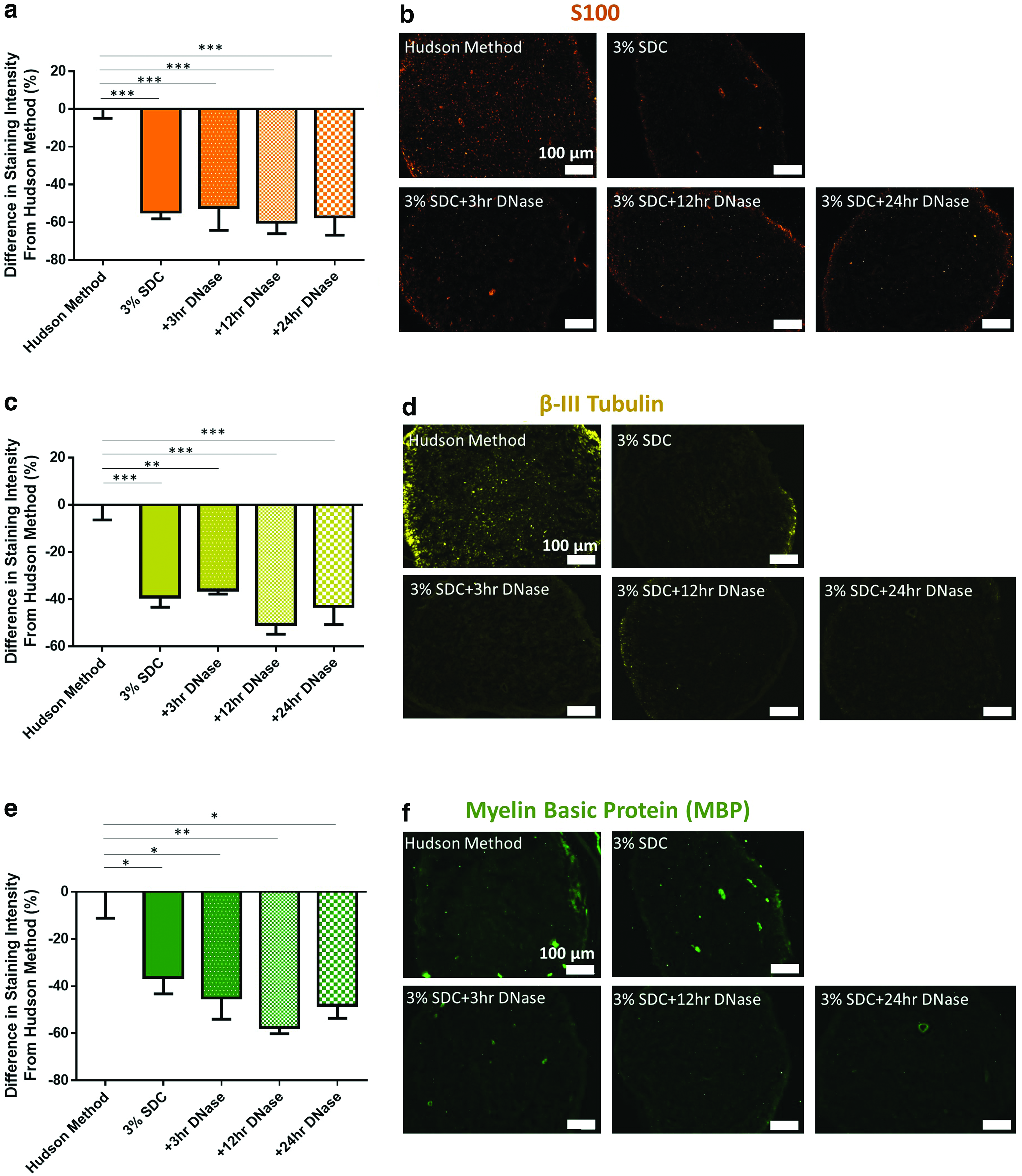
ECM preservation was also analyzed to ensure that additional incubation did not further compromise ECM content and structure. Collagen-I analysis revealed that the addition of any duration of DNase incubation did not significantly compromise collagen-I content compared with the 3% SDC (no DNase) decellularization process and the Hudson method (Fig. 5a, b). In addition, increasing the incubation time in DNase did not significantly alter the collagen-I content. However, basal lamina constituents, laminin and collagen-IV, were found to be affected by DNase incubation time (Fig. 5c–f, respectively). Compared with the 3% SDC process, adding a 24-h DNase incubation resulted in significant removal of laminin (p < 0.05) and collagen-IV (p < 0.05). These results demonstrate that DNase incubation time should be minimized to avoid basal lamina removal.
Selection of final optimized peripheral nerve chemical decellularization method
Based on these data, 3% SDC +3-h DNase was selected to replace Triton X-200 within the Hudson method. This modification of the Hudson method is now referred to as the SDD method and nerves decellularized in this process were further investigated via proteomic profiling and cytotoxicity assessment.
Proteomic profiling and analysis
Upon comparison of global proteome of nerves decellularized using the Hudson and SDD methods (Fig. 6a), only six detected proteins were significantly different (p < 0.05). These proteins are described in Figure 6b. Further analysis was conducted to separate individual proteins into their cellular locations (Fig. 6c, d). The Hudson method and SDD decellularized nerves had a significantly lower number of total spectral counts compared with fresh nerve controls (p < 0.05) (Fig. 6c). There was a marked reduction in cytoplasmic (p < 0.05), plasma membrane (p < 0.05), and nuclear-related proteins (p < 0.05) compared with fresh controls (Fig. 6c, d). Overall, the optimized SDD decellularization produces a strikingly similar global proteomic profile compared with Triton X-200, with only 1.67% of detected proteins being significantly different between decellularization techniques.
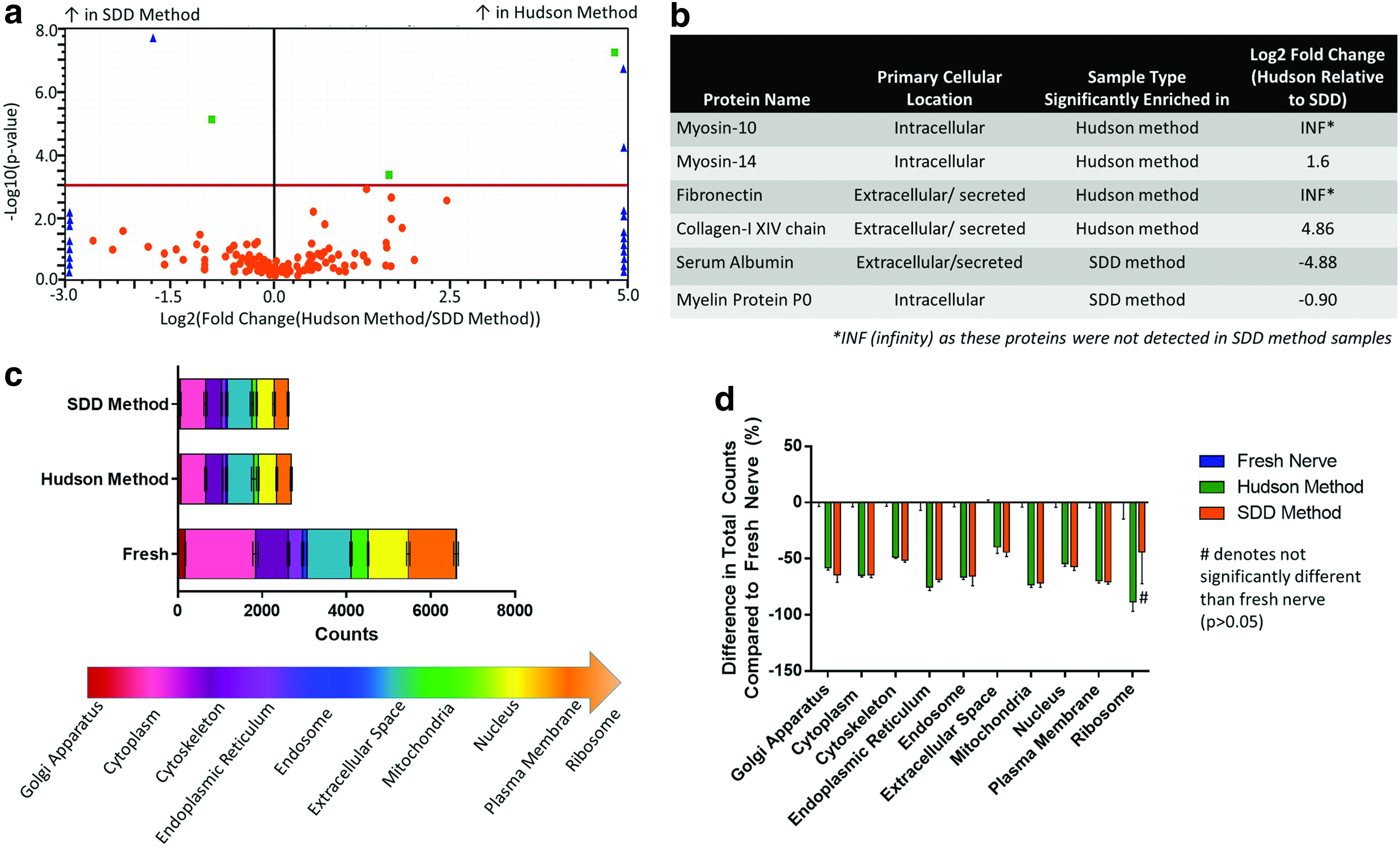
Validation of proteomics via ECM-targeted Western blotting
To validate the proteomic results, Western blotting for key ECM proteins within the Hudson method and SDD method decellularized peripheral nerves was also conducted (Supplementary Fig. S1a–c). Analysis of the electropherograms from the Western blots for laminin indicated no significant differences in relative quantity of this protein in the Hudson method versus SDD method decellularized nerves (Supplementary Fig. S1a). Collagen-I and collagen-IV Western blots and analysis also indicated no significant differences in relative quantity between the Hudson method and SDD method decellularized nerves (Supplementary Fig. S1b, c). These results help support and validate the findings made through semiquantitative IHC and proteomic analyses. Overall, Western blotting and analysis suggest that the SDD method at least matches the ECM preservation of the Hudson method.
Cytotoxicity assessment
An elution study was conducted on nerves decellularized using the Hudson method and SDD method to ensure that no cytotoxic residuals were present in the scaffolds (Fig. 7a). The conditioned media from the SDD decellularized nerves supported greater cell metabolic activity (80.95% ± 7.64) compared with that from the Hudson method decellularized samples (60.59% ± 11.44) (p < 0.05). As SDD method conditioned media surpassed the Hudson method conditioned media, the product of the SDD method of decellularization was determined to be cytocompatible and contained minimal cytotoxic residuals.
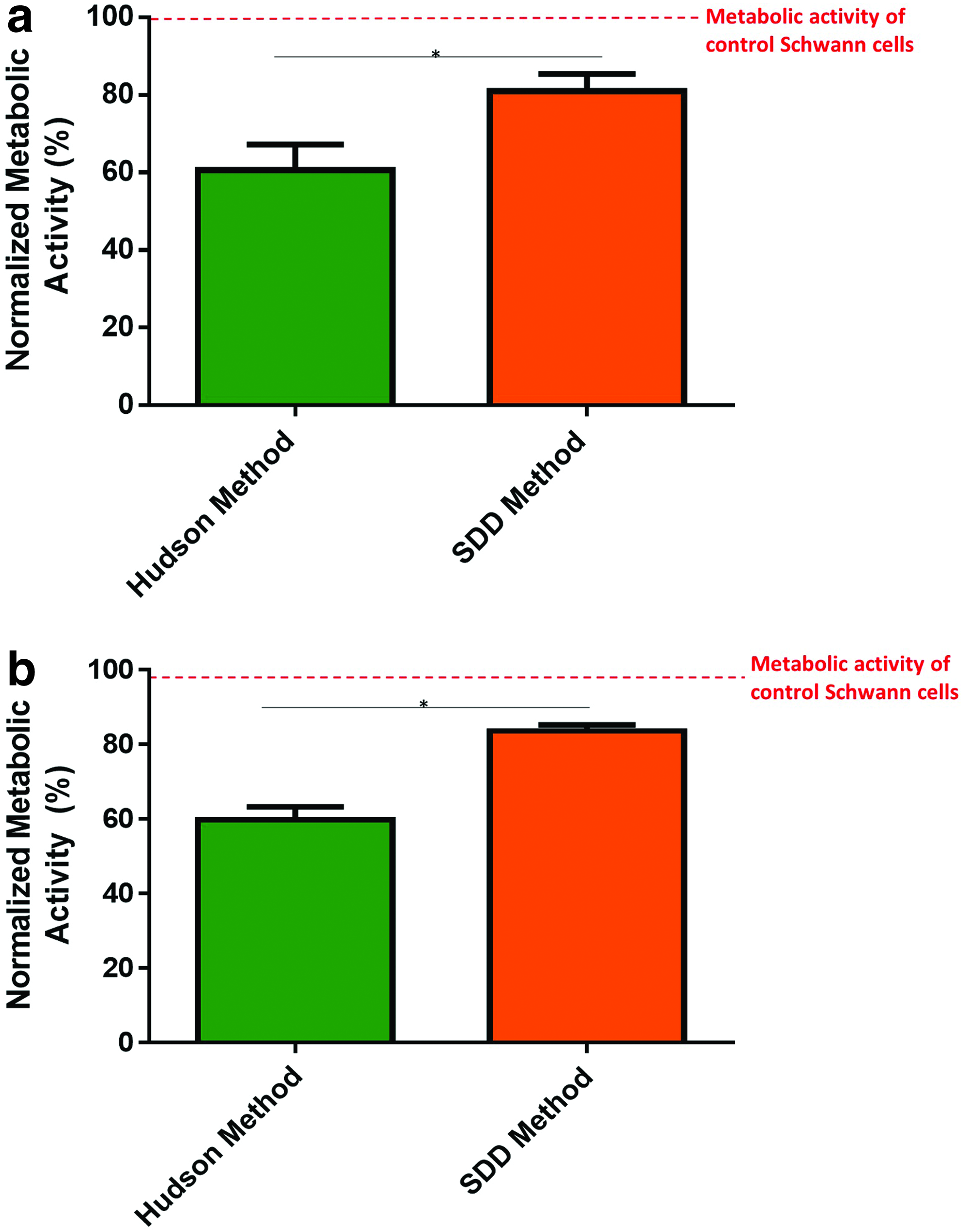
To ensure that no detergents were trapped within the scaffolds, in addition to the elution study, nerves decellularized using the Hudson method and SDD method were digested and cultured with Schwann cells (Fig. 7b). Similarly to the elution cytotoxicity results, SDD method homogenates supported greater cell metabolic activity (83.62% ± 2.82) compared with the Hudson method homogenates (59.87% ± 5.88) (p < 0.05). Together, these results demonstrate that SDD method decellularized nerves do not contain cytotoxic detergent residuals.
To better understand the cytotoxic threshold of SDC on Schwann cells, cells were exposed to increasing concentrations of SDC (Supplementary Fig. S2a, b). Results demonstrate that concentrations over 0.0015% SDC result in significant and substantial reduction in metabolic activity (<8.217% activity) and cytotoxic to Schwann cells. No difference in metabolic activity was seen between 0.015% and 1.5% SDC solutions, suggesting comparative cell death and dysfunction across these concentrations. The 0.0015% SDC exposure significantly reduces metabolic activity compared with control Schwann cells (74.32% ± 12.96%), however, the average metabolic activity is above 70% (set limit of healthy cell behavior). This concentration of SDC represents the cytotoxic threshold of SDC before inducing extensive cell death. These results suggest that the SDD decellularized scaffolds contain <0.0015% SDC and are cytocompatible.
Discussion
In this study, we aimed to develop a novel, optimized chemical decellularization method for peripheral nerve, which at least matches the Hudson method in terms of cellular removal and ECM preservation. In turn, an optimized decellularization process provides researchers with a reliable decellularization method for nerve tissue that uses the commercially available detergent SDC.
In literature, the two most widely used methods of chemical decellularization of peripheral nerve are the Sondell method and the Hudson method. In the Sondell method, nerves are subject to repeated water, Triton X-100, and SDC washes.27,32 This method has been shown to be effective at removing most myelin and other cellular components.17,26,32–34 Unfortunately, this method has also been demonstrated to significantly compromise the ECM, in particular the endoneurium laminin content and organization.17,26,32,34
The Hudson method of decellularization is an optimized method that uses repeated washes in buffer, zwitterionic detergents SB-10 and SB-16, and anionic detergent Triton X-200. 17 This method was demonstrated to have at least equivalent removal of cellular components and debris as the Sondell method17,34 while improving preservation of laminin and the basement membrane.17,34 This improvement in laminin preservation may be responsible for observations of greater cell adhesion to these acellular scaffolds 34 and improved regeneration. 35
As SDC was a key detergent in the Sondell method, SDC was also a chemical detergent investigated during the initial experiments of Hudson et al. in 2004 to determine an optimized decellularization protocol. 17 Hudson et al. found that while SDC facilitated superior removal of cellular components, the structure and content of the ECM were significantly compromised. 17 The Sondell method and Hudson et al. applied SDC at 4% for 24 h.17,32 In other tissues, SDC has been used to successfully decellularize a variety of tissues, including small intestine, 36 lung,37,38 liver, 39 and spinal cord. 40 For these tissues, the SDC concentration ranged from 1% to 4% (w/v) with washes ranging from 1 to 4 h at room temperature.36,38–45 While the concentration used by the Sondell method and Hudson et al. 35 was similar to literature on other tissues, the wash duration was 5–20 times longer.
This extended wash in SDC may be responsible for the excessive ECM removal. Therefore, in this study, reduced wash times that more closely match those in literature (2 and 1.5 h) were used. Furthermore, to optimize SDC concentration for peripheral nerve decellularization, 1%, 3%, and 5% SDC were investigated in this study.
Overall, implementing 3% SDC in place of Triton X-200 within the Hudson method facilitated greater axonal, Schwann cell, and myelin removal. The current study demonstrates that even at shorter wash times, SDC is still a potent remover of cellular components. Semiquantitative IHC analysis demonstrated that decellularization using the 3% SDC process did not cause excessive removal of ECM components collagen-I, collagen-IV, and laminin as staining was not significantly different from the Hudson method decellularized samples. Taking the ECM preservation into account, 3% SDC was the most optimal concentration of SDC to decellularize nerves as it removed cellular components comparably with 5% SDC, while sparing the scaffold unnecessary damage overall.
One issue that remained with SDC decellularization was remaining DNA debris. In all SDC (no DNase) decellularized samples, a bright and diffuse DAPI staining was observed. While this pattern of DNA staining indicates no visible nuclei, DNA is still present within the scaffolds. In other reports of SDC-based decellularization, a similar phenomenon was observed.2,46–49 The DNA may associate with the ECM via electrostatic interactions or agglutinate on the surface of the scaffolds.2,46,47
Differences in detergent exposure (24 and 18 h for Triton X-200 vs. 2 and 1.5 h for 3% SDC) may be responsible for these differences. In the Hudson method, nerves are exposed to Triton X-200 for 24 h and then again for 18 h. When SDC is implemented at similar times to Triton X-200, Hudson et al. demonstrated that while SDC had better cellular removal, it also caused excessive ECM damage. 17 To preserve the ECM, exposure time to SDC should be significantly decreased. In literature, SDC is typically applied for 1–4 h36,38–45 rather than 24 h.17,32 In this shorter exposure time to minimize ECM damage, the cellular DNA removal is lower thus requiring the use of enzymatic removal of DNA. Instead of increasing detergent exposure to remove nuclear debris, ECM damage was easily mitigated by implementing DNase incubations and salt buffer washes.
To facilitate DNA debris removal, incubation in DNase is commonly used in preclinical decellularization strategies.1,49,50 DNase enzyme is also used as part of some commercial decellularization processes.51,52 Inclusion of enzyme degradation, including DNase and ChABC, within a decellularization process does not necessarily compromise its Food and Drug Administration (FDA) status as minimally manipulated human cell, tissue, and cellular- and tissue-based product (HCT/P) if the scaffold is used, in this case, for peripheral nerve application (homologous use as autologous tissue). 53 For example, DermACELL® (LifeNet Health) is a decellularized human dermal tissue scaffold which undergoes decellularization that includes endonucleases but is classified as HCT/P by the FDA.51,54
If scaffolds generated from the SDD method of decellularization are utilized for applications other than peripheral nerve regeneration or are further processed, for example, to a hydrogel form, and applied to SCI similarly to Cerqueira et al. 10 and Cornelison et al., 11 it is likely the scaffolds would need to proceed through traditional medical device pathways, which may be more difficult than HCT/P pathways. However, evaluation by the FDA would be necessary for a concrete designation.
Like detergent steps, incubation in DNase needs to be optimized to ensure minimal damage to the ECM. In this study, we optimized DNase incubation duration by studying incubations ranging from 3 to 24 h. Staining and dsDNA quantification demonstrated that addition of DNase incubations as short as 3 h significantly reduces DNA content compared with both the 3% SDC (no DNase) decellularization process and the Hudson method. Additional DNase incubations did not further remove other cellular components but removed important basal lamina constituents if incubations approached 24 h. This investigation indicates that 3% SDC used in conjunction with a 3-h DNase incubation (SDD method) is an optimal replacement for Triton X-200 within the Hudson method to decellularize peripheral nerve.
Semiquantitative analysis of staining intensity is a simple and relatively quick method to probe for cell removal and ECM preservation. However, more quantitative analysis of global protein content should be conducted to validate semiquantitative data. Proteomic profiling by label-free quantitative mass spectrometry enables a more robust and quantitative assessment of protein content within decellularized scaffolds, and has been increasingly utilized to characterize decellularized scaffolds.37,55,56 However, with hundreds of proteins detected in a sample, data interpretation and presentation remain a challenge.
In this study, volcano plots were used to quickly identify significant differences in proteomic profiles generated by both the decellularization methods. To better understand removal of cellular components, proteins detected can be grouped based on their cellular location(s) and compared. Between the two methods of decellularization, we can observe strikingly similar profiles and removal of intracellular proteins from a given location, again supporting the claim that the SDD method is a comparable decellularization method with the Hudson method. Semiquantitative IHC and proteomic results were further supported by Western blotting and analysis for key ECM proteins collagen-I, collagen-IV, and laminin that suggested similar levels of these proteins in decellularized nerves from both methods.
Interestingly, the proteins determined to be significantly different in the proteomic analysis did not align with the significant differences observed in IHC. One potential source of difference in these methods is sample preparation. For proteomic analysis, whole decellularized peripheral nerves were solubilized. This contrasts with staining methods, where the tissues are sectioned and slides are stained for the proteins of interest.
Solubilizing the whole nerve allows for investigation of the graft in its entirety and may better represent the contents compared with representative tissue sections. For example, it is possible that removal from the bulk of the nerve scaffold occurred (as seen in IHC), but the protein was either incompletely removed from the edges of the scaffold or protein agglutinated on the surface of the graft. This highlights the need to analyze protein removal and preservation in decellularized tissues utilizing multiple methods. Here, the SDD method of peripheral nerve decellularization was shown to at least match the Hudson method of nerve decellularization regardless of assessment measure.
In this study, a variation on the International Standard Operations 10993 was used as a preliminary test of cytotoxicity of the decellularized scaffolds. 57 This cytotoxicity test shows whether any detergent residuals are present that may ultimately impact in vivo compatibility. Here, supernatants from the Hudson method decellularized scaffolds promoted less cell activity than the supernatants from the SDD decellularized scaffolds. Decellularized scaffolds generated using the Hudson method have been tested in vitro and in vivo for their biocompatibility and ability to support cell survival/growth.10,11,35 As SDD decellularization resulted in higher cell activity compared with a proven biocompatible scaffold, we believe scaffolds generated using the novel SDD process do not contain significant cytotoxic residuals.
To further ensure that detergents were not trapped within the scaffolds, Hudson method and SDD method scaffolds were homogenized and the resulting digest cultured with Schwann cells. SDD method homogenates supported cell activity better than Hudson method homogenates. These findings match the trend observed in the elution study. Based on these results and by estimating the cytotoxicity threshold for SDC on Schwann cells, the authors estimate that the scaffolds contain <0.0015% (w/v) SDC. Overall, cytotoxicity testing in this study suggests that the SDD method produces noncytotoxic decellularized peripheral nerve scaffolds.
Future work includes assessing the biocompatibility of these decellularized scaffolds and evaluating their use for in vitro and in vivo applications. In vitro, these decellularized peripheral nerves could be used to generate the next generation of in vitro models of peripheral nerve and peripheral nerve injury. In vivo, this material could be investigated as a therapeutic intervention for neural injury repair.
Conclusion
In conclusion, the novel decellularization method (SDD) that replaces Triton X-200 with 3% SDC in conjunction with a 3-h DNase incubation within a decellularization process is a viable replacement for the previously validated Hudson method 17 after the discontinuation of the key detergent Triton X-200. This method generates decellularized peripheral nerve scaffolds, which adequately removes cellular components while preserving the ECM content and structure. Decellularized scaffolds generated from the SDD method represent a class of ECM-rich material with a wide field of application both in vitro, as a culture or test bed substrate, and in vivo as a regenerative scaffold.
Footnotes
Acknowledgments
The authors thank Dr. Kari Basso with the University of Florida Mass Spectrometry Research and Education Center for her assistance and expertise with mass spectrometry, data analysis, and interpretation. The authors also thank the University of Florida's Interdisciplinary Center for Biotechnology Research Monoclonal Antibody Core for assistance with automated Western blotting.
Disclosure Statement
No competing financial interests exist.
Funding Information
Funds from an endowment with the University of Florida were used for this study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.