
Abstract
Neuromuscular junctions (NMJs), specialized synapses between motor neurons and muscle fibers, are essential for muscle activity. A simple and reproducible cell-based in vitro NMJ platform is needed to test the impact of chemicals on the neuron-muscle communication. Our platform utilizes genetically modified neurons and muscle cells, optimized culture conditions, and commercially available multielectrode array system for recording action potentials. Neuronal cells (NSC34) were optogenetically modified with channelrhodopsin chimera to allow for simultaneous, light-mediated, millisecond-precise activation of neuronal population. This signal is propagated through functional synapses to the muscle fibers. Muscle cells (C2C12) were modified by incorporating gap junction protein (Connexin-43) to improve intracellular communication without affecting muscle differentiation. This communication between muscle fibers resulted in better signal propagation and signal strength. Optimized culture medium facilitated the growth and differentiation of both cell types together. Our system was validated using vecuronium, a muscle relaxant, which abolished the muscle response. This in vitro model provides a unique tool for establishing a NMJ platform that is easy to record and analyze. Potential applications include nondestructive long-term screening of drugs affecting the NMJ.
Impact statement
A neuromuscular junction (NMJ) in vitro system is a useful tool to measure the effect of chemicals on the motor neuron—skeletal muscle connection. Here, we describe the use of genetically modified cells and optimized culture medium to establish a reliable and simple analytical system. Nondestructive neuronal stimulation and easy recording of electrical response has a broad range of applications including faster long-term screening of drugs affecting the NMJ.
Introduction
The nervous system, via peripheral neurons dictates and directs the response of muscles in the human body. Understanding this communication between neurons and muscle cells is key to neuromuscular research. However, this presents a tremendous challenge due to our inability to easily and accurately simulate in vitro neuron muscle interfaces or neuromuscular junctions (NMJs) that recapitulate physiological responses. Neurons and muscle cells communicate through synapses between these two cell types. The cells are never in direct contact. The NMJ, synapse between motor neurons (MNs) and skeletal muscles, is one of the most important connections within the human body. There is a need for high fidelity reliable in vitro NMJ systems that allow for simultaneous and nondestructive neuronal activation and simplified recording from contracting muscle cells.
Current in vitro NMJ models include simple two-dimensional (2D) coculture systems,1–5 2D compartmentalized coculture models,6–8 and three-dimensional (3D) structures9–12 and have been shown to produce synapses structurally similar to an in vivo NMJ; 13 and biological hallmarks of the synapse are present. 9 Compartmentalization of MNs and muscles, and generating 3D platforms while simulating an in vivo environment with higher fidelity add complexity to the NMJ platform resulting in lower throughput, and increased costs and time.
Growth and differentiation culture media are important requirements for proper development and maintenance of the skeletal muscles, MNs, and the subsequent NMJ formation. Current NMJ coculture models present several challenges; for one, the MNs and muscle are derived from different in vivo environments and when cultured separately have distinct and often incompatible culturing requirements. Second, many NMJ platforms rely on primary cell types isolated from rodents,2–4,9,14 MNs differentiated from embryonic or induced pluripotent stem cells1,5,10–12, or use muscle cell lines.5,7,12 Developing an NMJ coculture with cells from disparate etiologies results in a low incidence of muscle innervation by the neurons.
In current 2D in vitro NMJ models, neurons and muscle cells are cultured separately. After development of myotubes (MTs), neurons are added to the differentiated muscle cells, wherein synapses develop randomly and often infrequently. 4 A relevant culture medium needs to simultaneously support myocyte differentiation into multinucleated MTs, differentiation of MN, and facilitate physical interaction of the muscle and neuronal cells allowing for the formation of a functional NMJ. To date, there is no such optimized media that allows for the coculture and co-differentiation of these very different cell types and subsequent formation of NMJs.
Finally, to monitor proper NMJ function MNs need to be stimulated, the signal transferred from MN, and a response from the skeletal muscle recorded. The activation of MNs is performed by electrical or chemical stimulation.4,6,7,11 The gold standard for electrical activation and recording is patch clamp, which activates a single cell, is destructive, and records only from a single point. 5
To address these challenges, we systematically designed a culture media that enables both growth and differentiation of muscle and neuronal cells. Additionally, the media was designed such that it substantially decreased the spontaneous activity of the muscles improving our ability to accurately record and distinguish compound/agent/factor-induced muscles responses. Simultaneous and repeated neuronal activation can be accomplished by incorporating channelrhodopsin into MNs. Channelrhodopsin is photosensitive and when incorporated into MN can easily be activated with specific light waves. 15 This MN activation is specific, temporal, noninvasive, and allows the possibility of repeated stimulation for long-term recordings. Channelrhodopsin has been used successfully in several in vitro NMJ platforms.10,12 Further, the introduction of the gap junction protein, Connexin-43 (Cx43) into C2C12 cells amplifies response upon neuronal activation, significantly increasing the propagation of the electrical signal.
Here we describe the use of the mouse neuronal cell line, NSC34 and myoblasts C2C12, in the development of a high-throughput platform. NSC34 have inherent morphological and physiological properties resembling MNs in development,16–18 while C2C12 myoblasts are a widely accepted mouse cell line used to study skeletal muscle differentiation.19,20 We established growth conditions using immortalized cell lines for consistency, and systematically designed a single coculture and co-differentiation medium for MT and MN. This protocol produces thick contractible MTs, innervation of MTs by neuronal cells and consequently efficient NMJ formation. Muscle action potentials (APs) were monitored and recorded using commercially available multielectrode array (MEA) system.21,22 The MEA allows us to record responses from a population of cells from multiple sites within the array. The recording is noninvasive and can be repeated over time. Our novel in vitro NMJ model system provides a reliable and simple analytical platform for noninvasive stimulation, and an easy readout that will enable rapid and accurate screening of compounds that manifest their activity or toxicity via NMJs.
Methods
Medium optimization
C2C12 cells were seeded at 40,000 cells/cm2 in growth medium (GM); Dulbecco's modified Eagle medium (DMEM; ATCC), 10% fetal bovine serum (FBS; ATCC), and 1% Penicillin-Streptomycin (Pen-Strep; ATCC)) into a 24-well cell culture dish. Cells were grown for 24–48 h in GM, and at 80–90% confluence the medium in each well was replaced with test media. NSC34 cells were seeded in GM at 2000 cells/cm2 in a separate 24-well plate. Cells were grown in GM 24–48 h, and at about 30% confluence the medium was replaced with a test medium to initiate differentiation. Experiments were run for 7 days, and media changes were performed every other day. The composition of tested media is described in Table 1.
Different Media Composition Used in the Experiments in Figure 1
B27, serum substitute; DIR, muscle differentiation medium; DMEM, Dulbecco's modified Eagle medium; FBS, fetal bovine serum; HS, horse serum; IGF-1, insulin growth factor-1; N2, neuronal supplelment 2; NIR, neuron differentiation medium.
Generation of C2C12-Cx43 cells and NSC34 C1C2 cell line
The vector (pMCK-Cx43) containing full-length rat Cx43 under the muscle creatine kinase (MCK) promoter, and G418R gene under SV40 promoter was kindly provided by Dr. Hans Reinecke (University of Washington, Seattle, WA 23 ). Recombinant DNA plasmid was delivered in C2C12 cells in Lipofectamine3000 complex following the manufacturer's protocol. Cell clones were isolated from a pool of cells surviving 1 mg/mL G418 and the selected stable clonal cell lines were analyzed for protein expression via immunohistochemistry and Western blot (Supplementary Fig. S1 and Supplementary Methods). Optogenetically active neurons were generated using lentiviral transduction with pLenti-CaMKIIa-C1C2-EYFP (gift from Karl Deisseroth, Addgene, Plasmid #3551924) packaged, and delivered in a vector virus as previously described. 25 The ChR expression is under calmodulin-dependent protein kinase II promoter enabling specific expression in excitatory neurons. Single clones were isolated via fluorescence cell sorting (FACS) on BD ARIA with the argon laser. Clonal cell lines with stable expression of green fluorescent fusion protein (GFP) marker fused to ChR2 protein were selected for the functional assays. Optogenetic stimulation was performed using 460 nm collimated light-emitting diode (LED) light source connected to Prizmatix Pulser for train generation at 2 Hz, 50 ms pulses of 10 trains, repeated 5 times during each experiment.26,27
Immunocytochemistry, imaging, and analysis
Cells were fixed with 4% paraformaldehyde (PFA; Electron Microscopy) permeabilized with 0.4% Triton-X-100 (Sigma); and 1% bovine serum albumin fraction V (Invitrogen, Waltham, MA) was used as a blocking agent. Primary antibodies to MF20, synaptic vesicle protein 2 (SV2) (1 μg/mL, Cat. no. MF20s, Cat. no. SV2; Developmental Studies Hybridoma Bank), beta-III Tubulin (1:1000, Cat. no. ab18207; Abcam), and Cx43 (1:100, Cat. no. 3512; Cell Signaling Technology) were added, and counterstained with AlexaFluor secondary antibodies. Nuclei were labeled with NucBlue. Acetylcholine receptor (AChR) were stained using α-bungarotoxin conjugated with AlexaFluor. C2C12 cell images were analyzed by cell density, and fusion index. The fusion index is the ratio of the number of nuclei (4′,6-diamidino-2-phenylindole dihydrochloride [DAPI] staining) in MTs (myosin heavy chain [MHC] staining) to the total number of nuclei present in the image. NSC34 cell images were analyzed by cell density and neurite length per cell. Each image was analyzed using Image J software (National Institutes of Health, Bethesda, MD) with Neurite Tracer plug-in. 28 For all conditions, 10 images from each well were analyzed and the value was averaged for each sample. Minimum three samples of each condition were completed. Kruskal–Wallis and Dunn test as a post hoc were performed on data.
Experiment
Coculture of NSC34 and C2C12 cells
C2C12 cells were seeded at 40,000 cells/cm2 in GM for 24 h, then medium was switched to C2C12 differentiation media, designated as muscle differentiation medium (DIR) for 3 days. On day 3 NSC34 cells were seeded at 2000 cells/cm2 on top of C2C12 cells in NBN2 medium. Media changes were performed every other day.
MEA recording
Cells were seeded on MEA chips, either in 60MEA200/30iR-Ti or 60-6wellMEA200/30iR-Ti arrays. Arrays were coated with poly-D-lysine and laminin. Before recording, the MEA chips were moved to the MEA2100 system (MultiChannel Systems) equipped with temperature control and allowed to equilibrate for 10 min before recording. The data were acquired using Multi Channel Experimenter (MultiChannel Systems) at a sampling rate of 20 kHz for 10 min at 37°C. Data were filtered using Butterworth band pass filter with 200 Hz cutoff frequency and threshold of 5 × standard deviation were set to minimize both false-positive and missed detection. A Kruskal–Wallis test was performed, and followed by Dunn's test as a post hoc pairwise comparison on the nine electrodes from each three independent experiments and significance was defined as p < 0.05.
Calcium imaging
Cells were seeded in 24-well cell culture plates. They were grown and differentiated as stated above. Intracellular calcium dynamics were visualized using Cal-590 AM dye (AAT Bioquest). The cells were incubated in 5 μM Cal-590 with 0.04% Pluronic F-127, 1 mM probenecid in Hanks and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer for 1 h before they were washed. Afterward, cells were placed on the microscope and fluorescence signals were recorded with CCD camera at a frame rate of 200 ms/frame. Vecuronium (VC) (2 μM) was added to the cells 5 min before recording. Experiments were performed minimum three times. ChR2 expressing cells and regions of interests (ROIs) on the C2C12 MTs were randomly selected for the analysis for C2C12-Cx43 mono- and coculture experiments and NSC34-ChR2 neurons in monoculture. The change in calcium dye intensity was analyzed using ImageJ software.
Results
Two in one: formulation of universal media for mouse NMJ cell-based model
To develop a cost-effective and reproducible NMJ model for large-scale screening of pharmaceuticals and countermeasures for organophosphate compounds we focused on the mouse cell lines: NSC34, for MNs and C2C12, for MTs formation. The NSC34 cell line is a well characterized model for investigation of neurotoxicity, cytoskeletal organization, and axonal transport.16,17,29,30 Mouse C2C12s are a well-established cell line for the study of myogenesis, muscle biology, and metabolism.19,20,31,32 Both of these cell lines are generally accepted as viable candidates for immature NMJ formation.
Formation of a robust and reproducible NMJ model is dependent on the coculture and differentiation of NSC34 and myocytes into MNs and MTs, respectively. A significant challenge in MN and MT co-differentiation is their dependence on divergent and often incompatible growth factors. For instance, MT formation is highly dependent on the availability of serum in the differentiation media while neural stem cells (NSC) exit the mitotic phase and initiate differentiation only in serum-depleted media. Here we performed a systematic evaluation of the media composition to find the optimal formulation for the co-differentiation of both cell types (Table 1). We evaluated efficient MT and MN development through fusion index for MT formation (Fig. 1A, B) and cell density correlated to neurite length per cell as markers of neuronal differentiation (Fig. 1E, F).

Differentiation and proliferation of C2C12
Each cell line has an optimal media formulation when grown individually. The optimal media for C2C12 differentiation into MT consisted of 2% horse serum (2% HS), insulin growth factor-1 (IGF-1), and all trans-retinoic acid (atRA) in a DMEM media background (Table 1 and Fig. 1, designated as DIR). Our initial approach in developing the formulation to sustain co-differentiation was a serial serum dilution through addition of various fractions of DIR to neuron differentiation medium (NIR) (Table 1). Reduction of HS to 1%, through DIR:NIR fraction, slightly increased the fusion index of MT possibly due to the two-fold reduction of C2C12 growth (Fig. 1A, B, designated M2A). Omission of atRA had a more pronounced negative effect on the MT formation measured as fusion index (Fig. 1B, designated M2E), while removal of IGF-1 from the differentiation media resulted in slow myoblast growth (Fig. 1A, designated M2C) and did not significantly affect the cell fusion index (Fig. 1B). NSC34 did not exit the cell proliferation stage and no significant neurite growth was observed even at the lowest serum concentration (HS 0.5%) in the presence of both IGF-1 and atRA (Fig. 1E, F, designated M2D).
Our second approach was the replacement of DMEM with neurobasal (NB) medium. This improved neurite outgrowth in the presence of various sera combinations, however, NSC34 proliferation doubled, thus inhibiting the MN maturation process (Fig. 1C, D, G, H). Therefore, our next step was substitution of serum with growth factor replacement formulations, designated B27 and N2. The serum replacement, N2 supplement, decreased NSC34 cell proliferation providing conditions for optimum neurite length while sustaining fusion index of C2C12 differentiating cells comparable to serum-containing media. Serum-free neurobasal media supplemented with IGF-1 and atRA provided optimum conditions for mitotic arrest and differentiation of NSC34 cells into MNs characterized by minimal cell density and maximum neurite length (Fig. 1E, F, designated NIR). Thus, the absence of serum significantly promoted differentiation of NSC into MN while sustaining MT formation (Fig. 1C, D, G, H, designated NBN2). This NBN2 medium represents the most favorable formulation required for MN and MT co-differentiation with the explicit purpose of optimizing in vitro NMJ formation.
Optimization of cell culture protocol for MN and MT co-differentiation
Although the NBN2 media provided optimal conditions for NSC34 and C2C12 co-differentiation into MNs and MTs, the lack of serum resulted in poor attachment of differentiated MTs to the extracellular matrix. To improve MT attachment we cultured the C2C12 cells in DMEM with 2% HS for 3 days before the addition of the NSC34 cells in NBN2 medium, at which point the MTs were semi-differentiated. This media switch protocol resulted in improved differentiation and attachment of the MT and prolonged the viability of the MT/MN coculture. As such, we have cocultured mature MNs and MTs for 9 days without any apparent MT detachment. Furthermore, due to the reduced content of electrolytes in the NBN2 medium, we did not observe spontaneous twitching of the mature MTs, typical for cells cultured in DMEM media. Applying a calcium sensitive dye, Cal590, we validated the lack of spontaneous membrane depolarization of muscle tissue membranes in NBN2 medium (Fig. 2C). This did not only improve MT attachment and prolong cell culture viability, it also significantly improved data analysis by reducing variability originating from spontaneous muscle twitching (Fig. 2B, C).
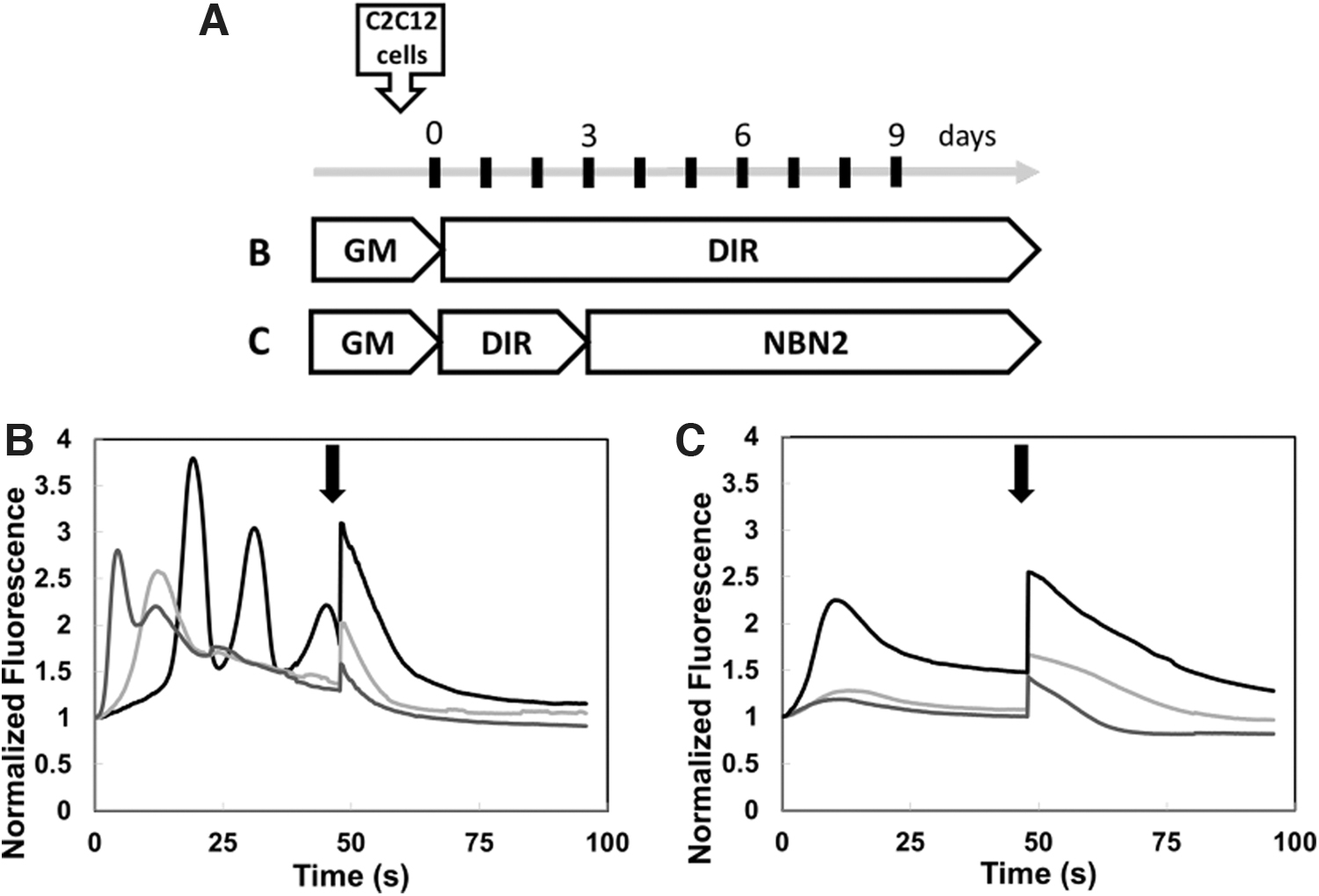
NBN2 medium decreased spontaneous muscle activity in C2C12 cells.
Engineering of cell-based NMJ assay with enhanced intracellular communication between muscle cells and noninvasive optical stimulation of MN
Noninvasive activation of MN will significantly improve the signal-to-noise readout and data interpretation of NMJ function upon addition of therapeutics. Optogenetics is a technique that allows for light-mediated, millisecond-precise activation of genetically modified neuronal populations. We generated a stable mouse neuronal cell line (NSC34 ChR) expressing light-gated ion channel protein, a chimera of channelrhodopsin 1 and 2 fused to GFP. Similar to wild-type MN, these transgenic neurons with a membrane-integrated ChR, expressed neuronal markers, SV2 and synaptophysin, upon differentiation into MN (Fig. 3). This engineered cell line is amenable to targeted, noninvasive activation of neurons.

NSC34 cells expressing channelrhodopsin chimera (ChR). The NSC34 ChR cells, upon differentiation into MNs, express neuronal markers: SV2 and Syn. DAPI stains nuclei. Scale bars, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride; MNs, motor neurons; SV2, synaptic vesicle protein 2; Syn, Synaptophysin.
However, on neuronal stimulation, the response from the muscle cells was weak and erratic likely due to the sparse density of NMJs that varied between experimental conditions. AChR clustering was increased when C2C12 cells were cocultured with MN, but this can vary between areas of the same plate or between experiments (Supplementary Fig. S2). One approach toward enhancing muscle response readout is synchronization of MT contraction. To ensure coordinated MT contraction in response to NMJ activation, we generated C2C12-based cell line harboring recombinant gene encoding for gap junction protein Cx43 under MCK promoter. 23 Expression of Cx43 from a MT specific promoter resulted in continuous accumulation of the gap junction protein during the myofiber maturation process and did not interfere with MT differentiation and fusion (Fig. 4 and Supplementary Fig. S1). Next, we applied a MEA to compare the AP generated by wild-type and Cx43-expressing MTs. We registered spontaneous AP from both cell types differentiated for 7 days in serum-free DMEM medium. As projected, the AP frequency, measured as spikes per sec, was increased in the muscle tissue expressing the gap junction protein compared to MT originating from wild-type myocytes. We registered no spontaneous AP activity from both cell types differentiated in NBN2 medium (Fig. 5).
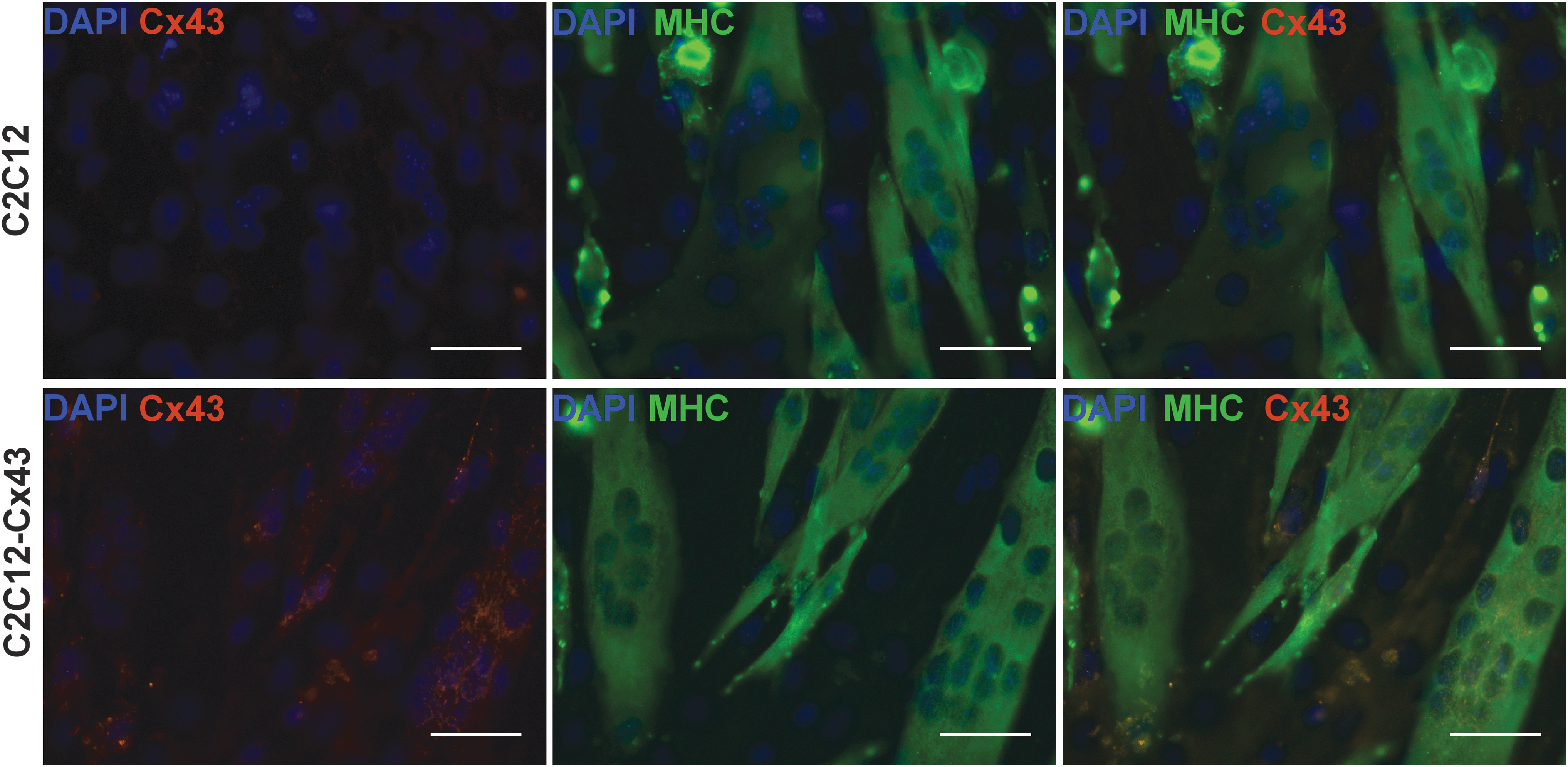
C2C12 cells express Cx43. Introduction of Cx43 under MCK promoter resulted in expression of Cx43 in differentiated MTs (bottom panels, arrows) not present in differentiated C2C12 MTs (top panels). MHC antibody stains differentiated MTs. DAPI stains nuclei. Scale bar, 100 μm. Cx43, Connexin-43; MCK, muscle creatine kinase; MHC, myosin heavy chain; MTs, myotubes.
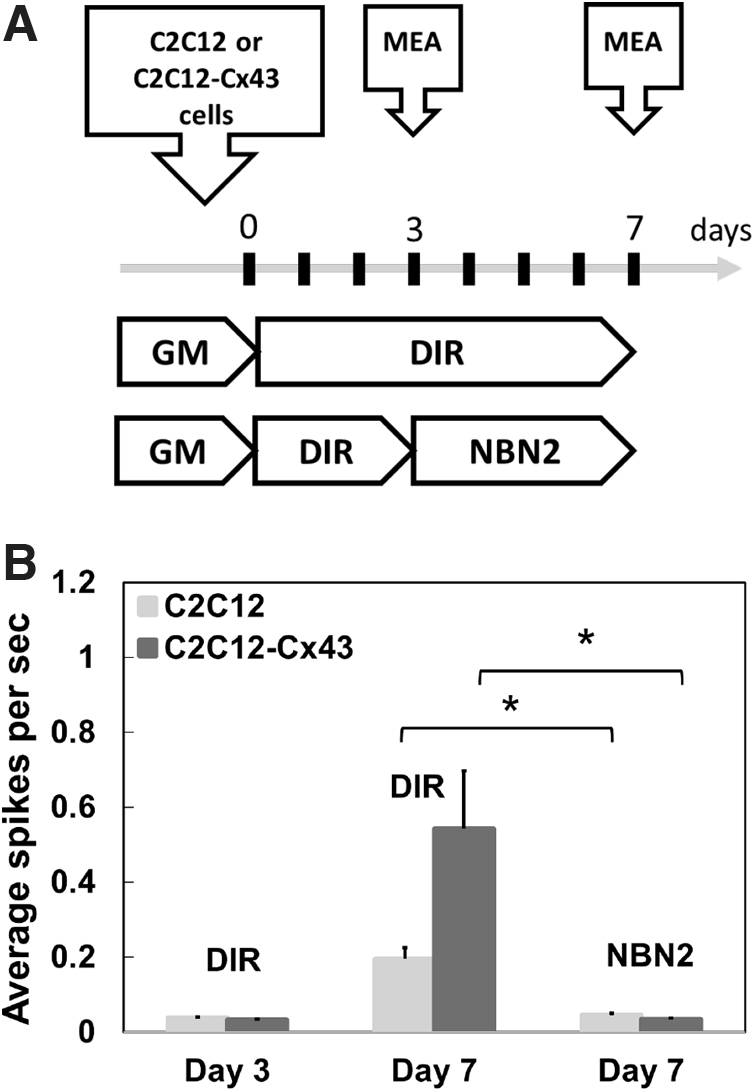
Cx43 modified muscle cells show higher AP than wt cells.
We validated the optogenetic control of NMJ function via two independent methods, electrophysiological measurement of AP on the MEA system and calcium influx imaging. We monitored Ca2+ transit responses upon LED exposure (at 460 nm) of MN or MT mono-cultures and cocultures of MNs and MTs. Fluorescent signal from the LED-triggered accumulation of intracellular Ca2+ was obtained from time-lapse microscopy of individual cells and was normalized to background fluorescence before stimulation. No light-induced Ca2+ transit was registered in MT mono-cultures, while MN originating from the NSC-ChR cells produced a consistent response to light stimulation (Fig. 6, LED panel). The normalized fluorescent signal from Ca2+ transit in MTs was markedly lower compared to MNs due to the indirect activation of muscle tissue through functional but low density NMJ. We further validated the functionality of NMJ via two tissue-specific inhibitors. VC, a muscle paralytic antagonist of the nicotinic acetylcholine receptor on the postjunctional membrane, had no effect on signal in MN monocultures while it abolished Ca2+ influx in muscle cells through NMJ block. This result is indicative of the formation of functional NMJ in our coculture model.
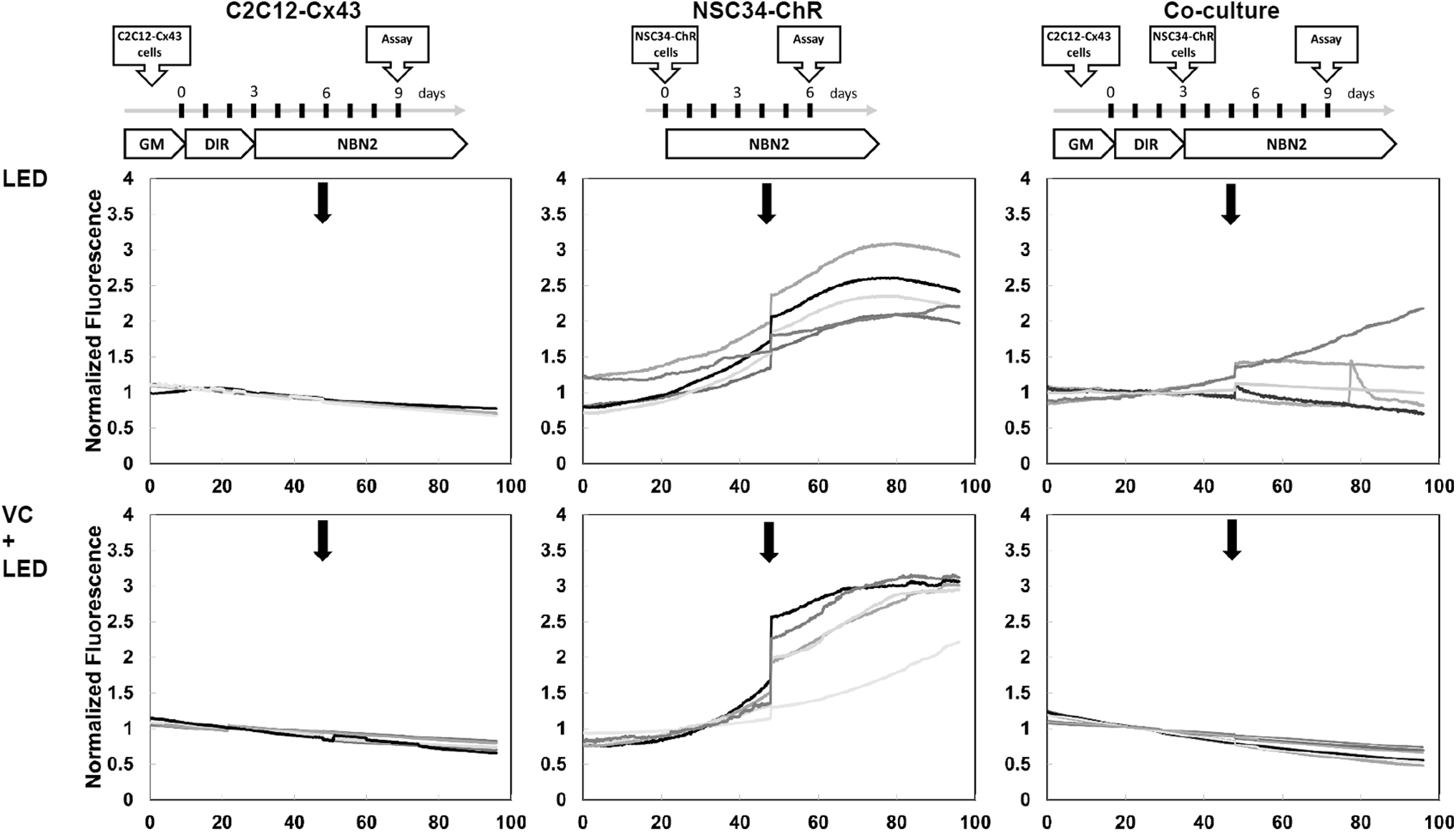
Functional NMJ formation. Calcium transit responses in monoculture and coculture conditions to optogenetic stimulation (2 Hz, 50 ms pulses of 10 trains, repeated 5 times during the experiment, LED, arrow). Muscle cells (C2C12-Cx43, left panel) did not respond to LED stimulation, however, when cocultured with MN cells (NSC34 ChR, right panel) an MN activated muscle response was observed, indicative of functional NMJs. VC, an antagonist of the nicotinic acetylcholine receptor (AChR), completely blocked the light-induced muscle calcium transits in all tested cocultures. VC did not have an effect on optogenetically induced activity of NSC34 cells. ChR2 expressing neurons and areas on MTs were randomly selected. Representative calcium transits from three independent experiments are shown. LED, light-emitting diode; NMJ, neuromuscular junction; VC, vecuronium.
To obtain quantitative data of NMJ activity, we measured cell membrane AP from mono and cocultures of MN and MT on the MEA system. As discussed previously, no spontaneous AP activity was detected before LED stimulation from all cell cultures differentiated in NBN2 media. In MN/MT coculture, the muscle cells were in direct contact with the electrodes as they were the first cells placed in the MEA (as outlined in Fig. 7A), therefore the recorded AP signal upon LED stimulation reflected largely muscle cell membrane depolarization through NMJ activation. MT mono-cultures did not mount APs upon LED stimulation and VC significantly inhibited the AP (Fig. 7B) indicating that the signal recorded by the MEA was, in fact generated by the muscle cells as a result of functional NMJs. The AP signal was enhanced in MN cocultures with MT expressing Cx43 compared to wild-type muscle tissue (Fig. 7B).

Functional NMJ formation.
In summary, we have developed a unique media formulation that allows for the coculture of MN and muscle cells facilitating the reproducible generation of NMJs in vitro. In addition, using optogenetically engineered MNs and Cx43 over-expressing MTs we were able to improve noninvasive neuronal stimulation and sensitive readout of NMJ function through synchronized muscle electrophysiological responses.
Discussion
Neurodegenerative diseases are often caused by dysfunctional activity of NMJ. While neuromuscular models enable a targeted understanding of disease models and rapid drug discovery, there are significant challenges in the development of NMJs in vitro. Herein, we describe a homologous in vitro coculture system capable of co-differentiation of myofibers and MNs, and generation of functional NMJs. Furthermore, incorporation of the light-gated ion channelrhodopsin allows us to noninvasively activate MNs and improve recording of electrical response from muscle cells using an MEA system.
The system presented here takes advantage of media optimization and cell modification for improved cell differentiation, neuronal stimulation, and muscle response recording. Both cell lines, C2C12 and NSC34 selected for this system required differentiation and needed to be cocultured to develop NMJ: myoblasts to MTs and neural stem cells to MNs. The natural dynamic of secretion of trophic factors is important for synaptogenesis, and many communication cues are lost when cells are matured and differentiated individually.6,33 Furthermore, muscle cells release factors and send cues to neurons that help them mature, enhance cell survival and neurite growth.6,33,34 MNs are also sources of signals that improve muscle differentiation, maturation, and contractile activities and contribute to NMJ maturation and stabilization. Our uniquely formulated co-differentiation media addresses the requirements of both cell types allowing them to differentiate together and form functional synapses. It facilitates synergism between neuronal and muscle cells resulting in optimal differentiation of and enhanced communication between the two cell types required for synaptic formation.33,34
Synchronized population activation is necessary for timely signal propagation and muscle responses. Incorporation of photoactivated ion channels into neurons is a well-established technique that enables the noninvasive activation of neurons using light and has been used in NMJ platforms.10,33,35–37 This cell-type-specific activation, allows for the exclusive stimulation of MNs without impacting the MTs in culture. Additionally, optogenetics offer the possibility of follow-up neuronal activation to check the rescue model. This cannot be accomplished using a patch clamp approach that offers only single-cell, one-time recording. Furthermore, chemical neuronal activation is not always specific: it can affect both neurons and muscles (e.g., KCl), affects different types of neurons in the mixed neuronal population (e.g., glutamic acid), or is an easily degradable compound resulting in response variability (e.g., Ach).
Skeletal muscles are excitable cells and generate electrical signals that can be recorded using an MEA. Electrical recording of extracellular APs was obtained from the primary myoblasts using a planar MEA21,22 and 3D electrodes. 38 The recordings were from spontaneously active muscles and not activated through an NMJ. The main challenge of electrical recording from muscles is the spontaneous activity of differentiated MTs that complicates the analysis of the induction through AChR and NMJ formation. Spontaneous muscle activity also leads to cell detachment and death and presents obvious challenges for subsequent studies. Our co-differentiation media substantially decreases the spontaneous activity allowing us to monitor AChRs-induced activity through an activated NMJ. This was confirmed in our experiments with AChR inhibitor, VC, which blocked muscle responses through induced NMJ activation. This allows us to predominantly monitor muscle activation through signals obtained from an NMJ as it occurs in fully developed muscle fibers. To increase electrical signals from muscle cells we took advantage of previously described data showing that grafted C2C12 cells expressing Cx43 were able to communicate with cardiomyocytes23,39 and considerably improved propagation of APs between both cell types in vitro 40 and in vivo. 41 Myoblasts express Cx4342,43 but downregulate the expression during differentiation into MTs. Introduction of Cx43 under muscle specific promoter also allows myoblasts to differentiate into MTs. 23 Recording was greatly improved by the usage of Cx43 modified cells (Fig. 7). Combining the ChR-expressing NSC34 cells with the Cx43-expressing C2C12 cells is a novel method to improve both excitation and signal propagation simultaneously.
A method of NMJ formation in vitro requires precise spatial control of muscle fibers by patterning, generation of trenches or 3D structures and guiding neuronal cells to muscle fiber using compartmentalized microfluidic devices.10,12 This approach is not feasible for large-scale screening of multiple compounds and their effect on NMJ synapse. 3D muscle structures and patterning help to establish more mature, adult-like muscle fibers,7,10,44 but co-differentiation of MNs and MTs also increased maturation of the muscle fiber in our 2D system. This demonstrates that not only structure but also intricate interaction between both cell types and time, play important roles in cell maturation and NMJ formation. 33 Also, patterning of muscle cells on MEA does not improve the signal propagation between MTs resulting in independent and non-synchronous MT contraction in the trenches, 45 similar to not fully developed MTs in short-term culture. 2D systems may be a simplistic approach but they are necessary for high-throughput drug discovery.
Conclusion
Here, we created a novel but simplified NMJ system that offers a rapid way to test multiple compounds efficiently and cost effectively as opposed to the currently available complex NMJ models.7,10–12 We envision the utility of this system as an initial screening tool for accelerated testing of therapeutics and toxins before in vivo animal studies and more complicated human-based platforms. Future validation of our system with known drugs affecting the NMJ connection and generation of drug dose responses will further prove its utility for screening purposes.
Footnotes
Acknowledgment
We would like to thank Dr. Hans Reinecke from Department of Pathology, University of Washington in Seattle for providing the vector (pMCK-Cx43).
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Defense Threat Reduction Agency (DTRA) interagency Agreement No. 1620298.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.