
Abstract
Fibroblast growth factor 23 (FGF23) plays a crucial role in managing renal phosphate and the synthesis of 1,25(OH)2-vitamin D3, which is essential for bone homeostasis. Developing robust in vitro systems to study FGF23-regulating mechanisms is crucial for advancing our knowledge and identifying potential therapeutic targets. The traditional in vitro 2D culture system results in relatively low expression of FGF23, complicating further exploration of its regulatory mechanisms and potential therapeutic targets. Herein, we reported a high-throughput approach to generate preosteoblastic cell spheroids with enhanced FGF23 production. For this purpose, murine preosteoblast cell line (MC3T3-E1) was cultured in our previously reported nonadherent microwells (200 µm in diameter, 148 µm in depth, and 100 µm space in between) and self-assembled into spheroids with a diameter of 92.3 ± 15.0 µm after 24 h. Compared with monolayer culture, the MC3T3-E1 spheroids showed a significant upregulation of FGF23 in both gene and protein levels after 24 h of serum-free induction. RNA sequencing and western blotting analysis further suggested that the enhanced FGF23 production in MC3T3-E1 spheroids was attributed to the activation of the parathyroid hormone (PTH)/PTH1R signaling pathway. Impressively, inhibition of PTH signaling through small molecular inhibitors or short hairpin RNA targeting PTH1R effectively reduced FGF23 production. In summary, the current study revealed the efficacy of the high-throughput formation of preosteoblast cell spheroid in stimulating FGF23 expression for mechanistic studies. Importantly, our findings highlight the potential of the current 3D spheroid system for target identification and drug discovery.
Impact Statement
This research introduces a high-throughput method for generating preosteoblastic spheroids that significantly enhance fibroblast growth factor 23 (FGF23) production via the parathyroid hormone signaling pathway. The findings offer a robust in vitro model for studying FGF23 regulation, addressing the limitations of traditional
Introduction
Fibroblast growth factor 23 (FGF23), a critical hormone primarily produced by osteoblasts and osteocytes in the bone, is essential in managing phosphorus, calcium, and vitamin D homeostasis. 1 It regulates phosphate reabsorption in the kidneys and inhibits the production of active vitamin D, thus maintaining the balance of phosphate and calcium in the body.2,3 Elevated levels of FGF23 are associated with multiple pathological conditions, such as chronic kidney disease (CKD), tumor-induced osteomalacia, and certain genetic disorders such as hypophosphatemic rickets.4–6 The pathological consequences of dysregulated FGF23 highlight the necessity of understanding the regulatory mechanisms governing its production. Developing robust in vitro systems to study FGF23-regulating mechanisms is crucial for advancing our knowledge and identifying potential therapeutic targets.
Traditional in vitro studies of FGF23 typically use 2D culture systems with osteocytic and osteoblastic cell lines, such as MC3T3-E1 and MLO-Y4 cells.7–9 While 2D cultures are relatively simple and cost-effective, they often result in very low levels of FGF23 expression and a limited response to stimulants, which does not accurately reflect the physiological conditions in vivo.10,11 One possible reason is that cells cultured in a 2D monolayer exhibited diminished mechanosensory and mechanotransduction capabilities, resulting in reduced mechanical stimuli that promote FGF23 synthesis. 12 Moreover, 2D cultures altered cell metabolic activity compared with the in vivo physiological state, which also affects the synthesis and section of FGF23.13,14 Furthermore, 2D cultures lack the hierarchical structure and cell–cell contact that are critical for the function of osteocytes and osteoblasts and FGF23 production. 15 Consequently, the regulatory mechanisms and potential therapeutic targets related to FGF23 cannot be effectively explored in 2D cultures.
To address the limitations of 2D cultures, researchers have explored cell-laden biomaterials that are archetypically seeded with individual cells and steered into the desired behaviors using exogenous stimuli to control growth and differentiation. 16 A previous study showed that the osteoblasts and osteocytes laden in Collagen I hydrogel exhibit elevated in vitro FGF23 production. 17 Furthermore, the encapsulated cells also gain a more stellate-like morphology and demonstrate a more robust response to parathyroid hormone (PTH) treatment and mechanical loading. 18 One study found that growing cells under 3D models produced more realistic mechanical effects and showed significant differences in FGF23 expression. 19 In addition, previous studies have also used a 3D bone tissue model to investigate FGF23 expression levels in bone cells, showing that 3D models more accurately reflect FGF23 expression levels. 20 However, the encapsulation of individual cells in biomaterials often creates the vast cell-free areas within the biomaterial, thereby lacking the direct cell–cell interactions necessary for the maturation of osteoblasts and osteocytes.21,22
In order to mimic cell–cell interactions, several cellular spheroids culture approaches, including centrifugation, low-adherence substrate, and hanging drop culture, have been proposed.23,24 However, the aforementioned approaches are not only laborious but also difficult for in vivo applications due to scalability challenges and uncontrolled complexity in 3D cell culture formats. 25 Furthermore, the underlying mechanism regulating the differentiation and maturation of cellular spheroids remains elusive.
We previously developed a high-throughput platform, which allows individual cells to self-aggregate into cellular spheroids containing 50–250 cells.16,26 Our previous work demonstrates the facile production of high quantities of skeletal progenitors, which exhibited enhanced differentiation capacities compared with the 2D monolayer counterparts. 26 Herein, we report the efficacy of the high-throughput formation of osteoblastic spheroids in stimulating FGF23 expression, which involves activation of PTH/PTH1R signaling through several key components, including the phosphorylation of protein kinase A (PKA) and cAMP response element-binding protein (CREB). Our findings highlight the potential of this 3D spheroid system in target identification and drug discovery for managing FGF23 production.
Materials and Methods
Cell culture
Mouse preosteoblast cell line (MC3T3-E1) was cultured at 37°C with 5% carbon dioxide (CO2) in alpha modification of Eagle’s medium (α-MEM) (HyClone, SH30265.01) supplemented with 10% fetal bovine serum (FBS; Gibco, No.2324371) and 1% penicillin–streptomycin (Cytiva, SV30010). When cell confluence reached 80%, cells were detached and 2 × 105/well seeded into plates. Cells were then incubated in α-MEM conditions for 24 h. For inhibitory experiments, MC3T3-E1 cells were pretreated with 10 µM H89 27 (Beyotime, S1643), diluted in dimethyl sulfoxide (DMSO) (PanReac AppliChem, A3672) to a final DMSO concentration of 0.1%. This pretreatment occurred 1 h before cell seeding. Subsequently, the cells were seeded and exposed to 10 µM H89 for 24 h. Controls were in 0.1% DMSO for 24 h. Cells were harvested for analysis after 24 h. For all in vitro experiments, unless otherwise specified, triplicates (n = 3) were used in each condition, and all the in vitro experiments were repeated for two runs.
High-throughput formation of MC3T3-E1spheroids
The expanded MC3T3-E1 was seeded in our previously reported agarose 24-well inserts containing ≈2000 microwells (Leijten et al. 16 ) at a concentration of 400,000 cells/well. Cells were cultured in serum-free medium at 37°C and 5% CO2 for 24 h. After that, the obtained spheroids were harvested by simple pipetting and centrifuged at 1000 g for 5 min for further analyses.
Live/dead staining
After 24 h of culture, cells cultured in monolayers (2D) and spheroids (3D) were removed from the culture medium and rinsed three times with phosphate-buffered saline (PBS, pH = 7.4). Then, live/dead staining was performed to assess the viability using Beyotime C2015S according to the manufacture’s instruction. Fluorescent images of live and dead cells were captured using a confocal microscope (Leica SP8; Leica) with excitation light of 490 and 545 nm, respectively. Images were analyzed using ImageJ software (ver. 1.51j).
Immunocytochemistry
Cell spheroids were collected from the culture plates and fixed with 4% paraformaldehyde. Thereafter, cells were permeabilized with 0.5% Triton X-100 in PBS and blocked with PBS containing 5% FBS and 0.1% Triton X-100. Next, Alexa Fluor 594-conjugated Phalloidin (UE, 1:100) in the blocking buffer for 2 h after three washes counterstained with Hoechst 33342 (Beyotime, 1:1000). Spheroids were then mounted with antifade (Invitrogen). Monolayer cells were grown on coverslips and stained using the same dilution ratio and protocol.
RNA sequencing and data analysis
Total RNA was extracted from spheroids and monolayer-cultured cells using the EZNA HP Total 19 RNA Kit (Omega). RNA quality was examined using an Agilent 2100 bioanalyzer. The bulk RNA sequencing (RNAseq) was conducted by the Beijing Genomics Institute (BGI). Single-stranded circular DNA libraries were prepared through the processes of polyadenylated RNA enrichment, reverse transcription, bubble adapter ligation, and polymerase chain reaction (PCR) amplification. Libraries were sequenced on the DNBSEQ platform by BGI. Raw data were filtered with SOAPnuke software to generate clean reads and mapped to the murine genome (GRCm38.p6) using HISAT software. Clean reads were then aligned to reference gene sequences using Bowtie. 28 Subsequently, the DESeq2 (v1.4.5) (www.bioconductor.org/packages/devel/bioc/html/DESeq2.html) software was used for data normalization and identifying differentially expressed genes between spheroids (3D) and monolayer-cultured cells (2D) with a Q value (adjusted p value) cutoff of 0.05. The volcano plot was created using the TBtools. 29 Kyoto Encyclopedia of Genes and Genomes (http://www.genome.jp/kegg/, accessed on January 1, 2022) pathway analyses were performed using the RNA Data Visualization System Dr. TOM system (BGI; http://www.bgi.com/global/dr-tom/).
Quantitative real-time PCR
The total RNA of cells was extracted with the EZNA HP Total RNA Kit (Omega) and reverse transcribed using HiScript III All-in-One RT SuperMix (Vazyme#R222). Quantitative reverse transcription PCR (qRT-PCR) was performed using the HiScript II One Step qRT-PCR SYBR Green Kit (Vazyme#Q711) on the CFX Connect Real-Time System (Bio-Rad#1855201). Primers used in this study were listed in Table 1, and the primers were synthesized by Aoke Biotech Co, Ltd. The sample size is five for each gene expression in each condition (n = 5). The expression of genes was quantified using Gapdh as a housekeeping gene, and the expressions of each gene were calculated using the 2−ΔΔCt method.
The Primers Used in This Study
Western blotting
Cells were lysed on ice in a lysis buffer (Merck Millipore) with a proteinase inhibitor (MedChemExpress, HY-K0010) and processed by centrifugation at 13,000 rpm for 10 min at 4°C. Protein concentrations were measured using a BCA Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. For each sample, 15 µg of denatured protein per well was loaded onto a 10% polyacrylamide gel, separated by electrophoresis, and then transferred onto nitrocellulose membranes (Millipore). After blocking with 5% nonfat milk in TBST for 1 h at room temperature, membranes were incubated overnight at 4°C with the following primary antibodies: rabbit anti-Phospho-PKA Substrate (1:1000; Cell Signaling Technology#9624S), rabbit anti-Phospho-creb1 (1:1000; Abclonal#AP0019), rabbit anticreb1 (1:1000; Abclonal#A11989), goat anti-FGF23 (1:4000; Immunotopic #186–206), and mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:8000; Proteintech#60004-1-I). On the next day, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Goat anti-Rabbit IgGAS014, 1:4000; or Goat anti-Mouse, AS071,1:4000) diluted in 5% nonfat milk for 1 h at room temperature. Signals were detected using WesternBright ECL solution (Advansta), and densitometry analysis that was standardized to GAPDH was performed with Fiji software. At least, triplicates (n = 3) were used for western blotting quantification.
Plasmid construction
In this study, we leveraged the GPP WEB PORTAL (GPP Web Portal—The TRC shRNA Design Process [broadinstitute.org]) for the design of shRNA sequences, prioritizing the ones with high predicted frequencies. The primers used in this study to construct the hairpin RNA (shRNA) targeting Pth1r are listed in Table 1. The shRNA sequences were cloned into the pLKO.1 puro vector (Addgene, #8453) after digestion with AgeI and EcoRI. The final construct, pLKO.1 shPth1r, was validated by sequencing to confirm the positive insertion of the shPth1r sequences.
Lentivirus production
HEK293T cells (Pricella, CL-0005) were seeded in 6-well plates at a density of 6 x105 cells per well and grown overnight. The following day, the cells were gently washed using PBS, followed by the addition of 0.5 mL of Dulbecco’s modified Eagle’s medium (DMEM) (Cytiva, SH30262.01) without FBS was added into each well. Simultaneously, a transfection mixture was prepared with 900 ng of psPAX2 (Addgene, 12260), 300 ng of pMD2.G (Addgene, 12259), and 1200 ng of shPth1r, along with 6 µL of Lipofectamine 2000 (Lipofectamine™, 11668027). This mixture was further diluted in 0.15 mL of OptiMEMTM (Gibco) and added to each well. The cells were incubated at 37°C for 8 h, after which 0.5 mL DMEM supplemented with 10% (v/v) FBS was replenished. The medium containing lentiviruses was harvested 48 h after transfection.
Lentiviral transduction of MC3T3-E1 cells
MC3T3-E1 cells were seeded in 6-well plates at a density of 1.5 × 104 cells/cm2. When the cells reached 70% confluence, they were transfected with either the experimental lentivirus (shPth1r) or the negative control (pLKO.1 shCon) lentivirus at 37°C for 8 h. Cells were selected with a selective medium containing 5 µg/mL puromycin (Beyotime, ST551) for 72 h to establish stable knockdown clones. After 4 days, the t stable clones were collected for subsequent experiments. The knockdown efficiency of Pth1r was confirmed using reverse transcription-quantitative PCR. Triplicates (n = 3) were used for the lentiviral transduction experiment.
Statistical analysis
All the numerical data were represented as mean ± standard deviation, and data were plotted as individual data points with bars representing the average value. Statistical analysis was performed using the GraphPad Prism software. Shapiro–Wilk test was used to test for normal distribution. A nonpaired unequal variance Student’s t-test or Mann–Whitney test was used for two-group experiments. For three-group comparisons, a one-way analysis of variance followed by a Bonferroni multiple comparisons posttest was applied. Statistical significance is indicated on all tables and graphs as follows: *p < 0.05. **p < 0.01, ***p < 0.001.
Results
High-throughput formation of osteogenic progenitor spheroids with elevated FGF23 production
The murine calvaria preosteoblast cell line MC3T3-E1 was seeded onto a 24-well plate containing microwells with 200 µm in diameter, 148 µm in depth, and 100 µm space in between (Fig. 1A). After 3 h, 200 cells were self-assembled in each microwell and formed a spheroid with a diameter of 92.3 ± 15.0 µm (Fig. 1B, C). The obtained spheroids remained stable during 24 h in vitro culture, and the average diameter decreased to 51.4 ± 5.8 µm after 24 h (Fig. 1B, C).

The high-throughput generation of murine osteoblastic progenitors (MC3T3-E1) spheroid enhanced osteoblastic differentiation and FGF23 production.
Live and dead staining revealed that the majority of cells within the spheroids remained viable after 24 h (Fig. 1D). Compared with monolayer cells, the MC3T3-E1 spheroids exhibited distinct cytoskeleton reorganization over 24 h in vitro culture (Fig. 1D). Furthermore, MC3T3-E1 spheroids exhibited a significantly higher expression of FGF23 than monolayer cells, with approximately a 10-fold increase in mRNA levels and a 2-fold increase in protein levels (Fig. 1E–G). Additionally, several osteogenic markers, including Ocn, Dmp1, and Sost, were also significantly upregulated (Fig. 1H–J).
The osteogenic progenitor spheroids enhanced PTH/PTH1R signaling pathway
To better explore the mechanism underlying the elevated FGF23 production of the osteogenic progenitor spheroids, we conducted RNAseq analysis of monolayer cultured (2D) and spheroids (3D) of MC3T3-E1. Compared with the monolayer (2D), MC3T3-E1 spheroids displayed dramatic gene expression changes, with a total of 1160 upregulated genes [
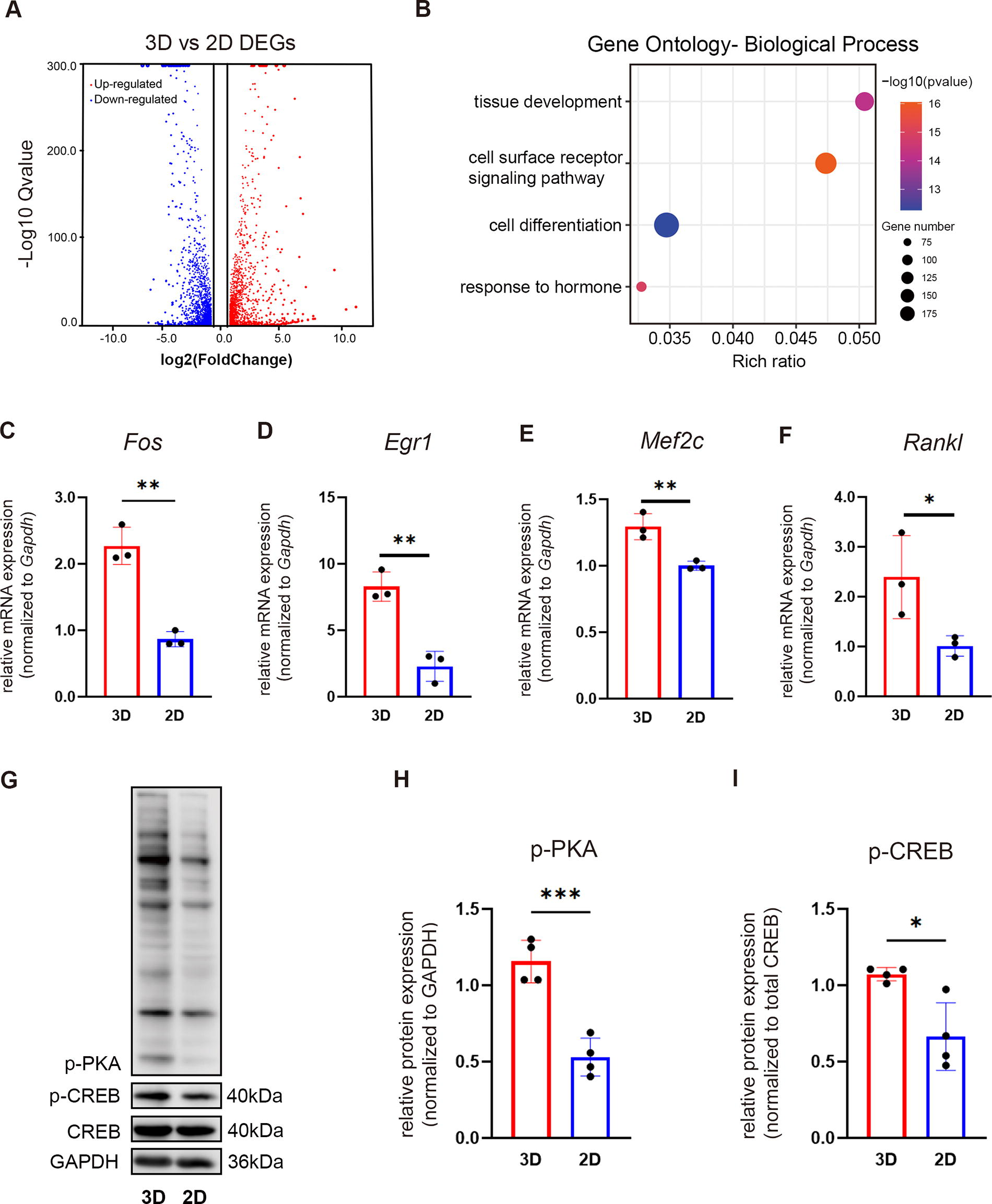
Murine osteoblastic progenitors (MC3T3-E1) MC3T3-E1 spheroids enhanced FGF23 expression via PTH signaling.
To verify the activation of the PTH signaling pathway in MC3T3-E1 spheroids, we performed western blotting on MC3T3-E1 spheroids and monolayer cells over 24 h in vitro culture in serum-free condition. The spheroids (3D) showed significantly higher levels of phosphorylated PKA (p-PKA) and CREB (p-CREB) than monolayer-cultured cells (2D) (Fig. 2G-I), suggesting that the FGF23 upregulation observed in spheroids was potentially regulated by the activation of the PTH/PTH1R signaling pathway.
Inhibition of PTH/PTH1R signaling attenuated FGF23 expression in MC3T3-E1 spheroids
To further verify whether PTH/PTH1R signaling mediates FGF23 expression in MC3T3-E1 spheroids, we knocked down PTH receptor 1 in MC3T3-E1 cells using lentiviral sh-Pth1r (Fig. 3A). Quantitative PCR data confirmed the sh-RNA mediated silencing of PTH1R was associated with an ∼60% reduction of Pth1r mRNA (Fig. 3B). Compared with the control group, the knockdown of Pth1r impaired the PTH/PTH1R signaling, evidenced by a significant decrease of p-PKA and p-CREB (Fig. 3C–E), in line with a significant downregulation of Fos, the PTH/PTH1R signaling target gene (Fig. 3F). Consequently, the sh-Pth1r group showed a decline in FGF23 expression in both mRNA (Fig. 3G) and protein levels (Fig. 3H-I).
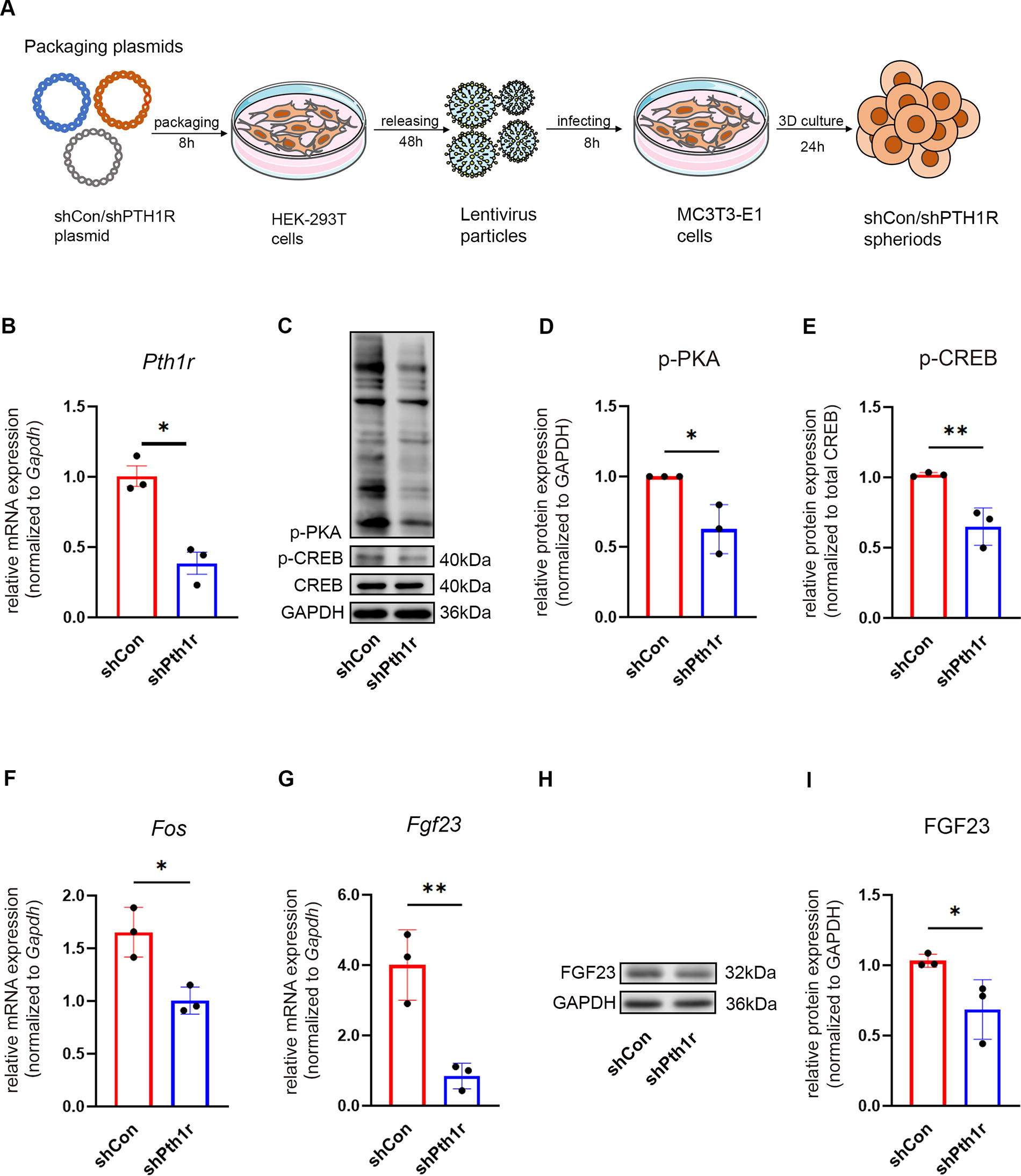
Knockdown of Pth1r attenuated FGF23 expression in murine osteoblastic progenitors MC3T3-E1 spheroids.
Next, we conducted a PKA kinase activity inhibition experiment to verify the influence of PTH/PTH1R signaling on regulating FGF23 expression in MC3T3-E1 spheroids. To this end, MC3T3-E1 in spheroids was cultured in serum-free conditions for 24 h and treated with or without the PKA inhibitor, H-89 (10 μM). Upon inhibition, MC3T3-E1 spheroids showed a significant decrease of p-PKA (up to 40%), although the phosphorylation of CREB remains unaffected (Fig. 4A–C). Consistently, the FGF23 expression in both mRNA and protein levels was also evidently downregulated in MC3T3-E1 spheroids upon inhibition of PKA kinase activity (Fig. 4D–F). These results demonstrate the regulatory effect of the PTH/PTH1R signaling pathway on FGF23 expression in MC3T3E1 spheroids (Fig. 5).

Inhibition of protein kinase A (PKA) kinase activity attenuated FGF23 expression in murine osteoblastic progenitors (MC3T3-E1) spheroids.

Schematic representation of the activation of PTHR1/PKA/CREB signaling elevated FGF expression in murine osteoblastic progenitors (MC3T3-E1) spheroids. Self-assembly of individual MC3T3-E1 cells into spheroids facilitates the activation of cell surface receptor PTH1R, subsequently leading to the activation of protein kinase A (PKA). Consequently, phosphorylated PKA increases the phosphorylation of cAMP response element-binding protein (CREB), and the transcription of osteogenic differentiation marker genes Ocn, Dmp1, and Sost, thereby upregulating Fgf23 expression. AC, adenylyl cyclase.
Discussion
FGF23 plays a crucial role in various fields, including skeletal biology, mineral metabolism, and CKD.30–33 Recent advancements indicate that reducing FGF23 signaling could mitigate these dental and skeletal anomalies, indicating the importance of understanding the regulatory mechanism of FGF23 production.34–36 Therefore, it is crucial to develop a robust in vitro system to study FGF23-regulating mechanisms and identify potential therapeutic targets for FGF23 disorders. The monolayer culture of osteocytic and osteoblastic cell lines has been widely used for in vitro FGF23 studies. However, the monolayer culture lacks the hierarchical structure and cell–cell contact, which is crucial for the function of osteocytes and osteoblasts; thus, the FGF23 expression in these systems is extremely low or even undetectable.37–39
In order to mimic the hierarchical structure within native tissue, numerous studies have explored 3D culture approaches including using scaffolding biomaterials or cell pallets generated by centrifugation or hanging drop techniques.31–34 However, the complex and labor-intensive procedures of the aforementioned approaches limit their large-scale application. In this study, we have employed a high-throughput approach to generate stable osteoblastic spheroids composed of a nonadhesive microwell array prepared by our previously established soft photolithographic technique. 16 Compared with the conventional 3D culture approach, our approach could generate versatile cellular spheroids composed of 200 cells in a high-throughput manner.16,26 In addition, the submicron-scale size of the obtained cellular spheroids facilitates the nutrient diffusion during in vitro culture and ensures cell viability and homogenous differentiation within the spheroids. 26
In this study, the MC3T3E1 cell spheroids showed a significant upregulation of FGF23 after 24 h in both gene and protein levels, indicating the maturation of the osteoblastic cells.40–42 Similar upregulation of FGF23 and Ocn was also observed in primary mouse periosteum derived skeletal stem/progenitor cell spheroids (Supplementary Fig. S1), suggesting the validity of this culture system on primary cells. We also explored the mechanism driving FGF23 upregulation. Our data revealed that the elevated FGF23 expression in MC3T3-E1 cell spheroids correlates highly with the elevated PTH/PTH1R signaling, which directly regulates osteoblast maturation and FGF23 production. 43 The exact mechanism driving the elevation of PTH/PTH1R signaling still needs further investigation. One possible explanation is that the mechanical cues within cell–cell and cell–matrix interactions thus subsequently enhance the endogenous ligand recognition and structural transition of a PTH receptor. 44 These findings indicate that targeting the PTH/PTH1R signaling pathway could be a potential therapeutic strategy for disorders involving abnormal FGF23 production. In light of this, our high-throughput method for generating osteoblastic spheroids offers significant advantages for target selection using small inhibitor libraries and CRISPR libraries.
In summary, in this study, we report a high-throughput approach to generate preosteoblastic cell spheroids with enhanced FGF23 production driven by the activation of PTH/PTH1R signaling. The scalability and efficiency of this approach allow for rapid screening and identification of potential therapeutic targets involved in FGF23 regulation. This capability provides a robust platform to explore and validate novel inhibitors or genetic modifications aimed at modulating FGF23 production. In addition, the reported high-throughput approach is not only limited to cell lines but also applicable to primary cells, which opens the possibility of screening the cells harvested from patients suffering from FGF23 disorders.
Footnotes
Acknowledgments
The authors would like to acknowledge Ms. Y. Zhang, Ms. Y. Zuo, and Mr. W. Yang for their great assistance in laboratory work. The authors would also like to appreciate the financial support from the National Natural Science Foundation of China, the Key Research and Development Program of Hubei Province, the Fundamental Research Funds for the Central Universities, and the ITI Research Foundation.
Author’s Contributions
J.J.: contributed to data acquisition and interpretation, performed all statistical analyses, and drafted and critically revised the article. J.Z., H.L., and S.J.: contributed to data acquisition and interpretation and critically revised the article. Q.H. and W.J.: contributed to conception, design, and data interpretation and drafted and critically revised the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.