
Abstract
Background:
Since many thyroid cancer tissue samples from atomic bomb (A-bomb) survivors have been preserved for several decades as unbuffered formalin-fixed, paraffin-embedded specimens, molecular oncological analysis of such archival specimens is indispensable for clarifying the mechanisms of thyroid carcinogenesis in A-bomb survivors. Although RET gene rearrangements are the most important targets, it is a difficult task to examine all of the 13 known types of RET gene rearrangements with the use of the limited quantity of RNA that has been extracted from invaluable paraffin-embedded tissue specimens of A-bomb survivors. In this study, we established an improved 5′ rapid amplification of cDNA ends (RACE) method using a small amount of RNA extracted from archival thyroid cancer tissue specimens.
Methods:
Three archival thyroid cancer tissue specimens from three different patients were used as in-house controls to determine the conditions for an improved switching mechanism at 5′ end of RNA transcript (SMART™) RACE method; one tissue specimen with RET/PTC1 rearrangement and one with RET/PTC3 rearrangement were used as positive samples. One other specimen, used as a negative sample, revealed no detectable expression of the RET gene tyrosine kinase domain.
Results:
We established a 5′ RACE method using an amount of RNA as small as 10 ng extracted from long-term preserved, unbuffered formalin-fixed, paraffin-embedded thyroid cancer tissue by application of SMART technology. This improved SMART RACE method not only identified common RET gene rearrangements, but also isolated a clone containing a 93-bp insert of rare RTE/PTC8 in RNA extracted from formalin-fixed, paraffin-embedded thyroid cancer specimens from one A-bomb survivor who had been exposed to a high radiation dose. In addition, in the papillary thyroid cancer of another high-dose A-bomb survivor, this method detected one novel type of RET gene rearrangement whose partner gene is acyl coenzyme A binding domain 5, located on chromosome 10p.
Conclusion:
We conclude that our improved SMART RACE method is expected to prove useful in molecular analyses using archival formalin-fixed, paraffin-embedded tissue samples of limited quantity.
Background
Since many thyroid cancer tissue samples from atomic bomb (A-bomb) survivors have been preserved for a long time as unbuffered formalin-fixed, paraffin-embedded specimens, analysis of such archival specimens is indispensable for clarifying the characteristics of thyroid carcinogenesis in A-bomb survivors. RET gene rearrangements are thought to play an initial and critical role in papillary thyroid cancer development, although wide variations in the prevalence of such rearrangements, ranging from 3% to 70%, have been observed in different geographic areas (2 –4). To date, at least 13 rearranged forms of the RET gene (RET/PTC 1–9, PCM1-RET, ELKS-RET, ΔRFP-RET, and HOOK3-RET) have been identified, of which RET/PTC1 and RET/PTC3 are by far the most frequent, from papillary thyroid cancers in patients both with and without radiation-exposure history (3,5 –7). Analysis of RET gene rearrangements is one of the most important issues in molecular thyroid cancer research among A-bomb survivors. However, it is a difficult task to examine all types of RET rearrangements because of the limited quantity of paraffin-embedded tissue specimens from A-bomb survivors; namely, it is almost impossible to investigate all 13 rearranged forms of the RET gene using such limited amounts of RNA.
Clontech's (Mountain View, CA) switching mechanism at 5′ end of RNA transcript (SMART™) technology is based on the terminal transferase activity of reverse transcriptase, which adds a few additional nucleotides, primarily deoxycytidine, to the 3′ end of cDNA, creating an extended template at the 5′ end in combination with a SMART oligonucleotide (8). Therefore, PCR amplification of cDNA using a set of adaptor-specific primer and gene-specific primer will produce the cDNA that contains the complete 5′ end of mRNA (9).
We improved and established the 5′ rapid amplification of cDNA ends (RACE) method (10) using a very small amount of RNA extracted from unbuffered paraffin-embedded thyroid cancer tissue by application of SMART technology. In addition to the analysis of common RET gene rearrangements, this method succeeded in detecting not only rare RET gene rearrangements but also one RET gene rearrangement that has not yet been reported.
Materials and Methods
Tissue
All cancer tissue specimens used for this study were prepared from archival unbuffered formalin-fixed, paraffin-embedded blocks. Three thyroid cancer tissue specimens from three different patients were used as in-house controls to determine the conditions for an improved SMART RACE method; one tissue specimen with RET/PTC1 rearrangement and one with RET/PTC3 rearrangement were used as positive samples. One other specimen, used as a negative sample, revealed no detectable expression of the RET gene tyrosine kinase (TK) domain. The control samples had been preserved at room temperature for 19–21 years. Papillary thyroid cancer specimens from A-bomb survivors, preserved at room temperature for 20–50 years, were collected after approval from the Human Investigation Committee and the Ethics Committee for Genome Research at the Radiation Effects Research Foundation (RERF). After deparaffinization of 5 μm sections by Hemo-De (Fujisawa Yakuhin Kogyo, Osaka, Japan) and staining with methyl green (Sigma-Aldrich, St. Louis, MO), cancerous regions were isolated using disposable scalpels. All cancerous regions from two to four successive tissue sections were combined for RNA extraction.
RNA extraction
RNA was isolated from dissected tissue using the High Pure RNA Paraffin kit according to the manufacture's instructions (Roche Diagnostics, Manheim, Germany), with some modifications. Briefly, dissected tissue was digested with proteinase K at 55°C overnight, followed by DNase I treatment. After the lysate was purified by High Pure filter, RNA was eluted twice with 100 μL of RNase-free water. RNA was then precipitated by ethanol in the presence of 2 μL of ethachinmate (Nippon Gene, Tokyo, Japan) as a carrier and resuspended in 30 μL of RNase-free water.
cDNA synthesis
To enhance template activity of RNA extracted from archival unbuffered formalin-fixed, paraffin-embedded tissue specimens, total RNA was first heated in 10 mM of citrate buffer (pH 4.0) at 70°C for 45 minutes and then precipitated by ethanol (11). Then, 100 ng of total RNA and 50 pmol/μL of random primers (9 mer) were heated in 11 μL of RNase-free water at 65°C for 10 minutes and chilled in ice water. A mixture consisting of 4 μL of 5 × reverse transcription (RT) buffer, 2 μL of 20 mM DTT, 1 μL of 10 mM dNTPs, and 1 μL of RNase Inhibitor (20 U/μL; TaKaRa, Tokyo, Japan) was added to the RNA solution and incubated at room temperature for 5 minutes. After addition of 1 μL of Rever Tra Ace (100 U/μL; Toyobo, Osaka, Japan), the reaction mixture was incubated at 42°C for 45 minutes.
Improved SMART RACE
After 1 μL of various concentrations of SMART adaptor (SMART II A oligonucleotide; Clontech) was added to 20 μL of cDNA solution, the mixture was further incubated at 42°C for 60 minutes and heated at 70°C for 15 minutes to inactivate reverse transcriptase. SMART RACE-PCR was performed in a 25 μL volume containing 1 × PCR buffer; 200 μM each of dATP, dCTP, dGTP, and dTTP; 3 mM of MgCl2; 0.4 μM of each primer (SMART adaptor-specific primer, S-RACE 1, 5′-AAGCAGTGGTAACAACGCAGAGTA-3′; exon 12 of RET gene-specific primer, RET-Ex12PR9, 5′-TCCGAGGGAATTCCCACTTT-3′); 0.5 U of FastStart Taq DNA polymerase containing a thermostable proof-reading protein (Roche Diagnostics); and 2 μL of SMART adaptor-treated cDNA. PCR was carried out on a DNA Engine using the following cycle conditions: at 95°C for 3 minutes; 45 cycles at 95°C for 30 seconds, at 60°C for 30 seconds, and at 72°C for 45 seconds; and a final extension at 72°C for 5 minutes. The first PCR products were diluted at 1000- to 10,000-fold in water, and 1 μL of diluted samples was used as a template for semi-nested SMART RACE-PCR with SMART adaptor and nested primer (RET gene-specific primer, RET-Ex12A4; Table 1). The second PCR conditions were as described in the first PCR, with the exception of annealing temperature (66°C instead of 60°C) and number of cycles (25 cycles instead of 45 cycles).
Cloning and sequencing of cDNA fragments
Five microliters of second SMART RACE-PCR products was electrophoresed on an 8% acrylamide gel and viewed with ethidium bromide. Target candidate bands were determined by BamHI (TaKaRa) digestion of aliquots of second SMART RACE-PCR products, because the fragments amplified by SMART RACE-PCR included a BamHI recognition site located at the 5′ end of exon 12 of the RET gene. Target candidate cDNA fragments derived from 50 μL of second SMART RACE-PCR products were eluted from 8% acrylamide gel and cloned into HincII-digested and dephosphorylated pUC118 vector using TaKaRa Blunting Kination Ligation kit (TaKaRa). Plasmid DNA containing a longer insert than 70 bp was sequenced using DNA sequencer CEQ8000 (Beckman Coulter, Fullerton, CA), because the total length of the SMART adaptor and the 5′ portion of exon 12 of RET was 55 bp.
Detection of RET/PTC8 expression in papillary thyroid cancer from A-bomb survivors by RT-PCR
Amplification of the BCR gene as an internal control by RT-PCR was at first conducted for examination of availability of RNA extracted from archival tissue samples. The cDNA derived from 10 ng of total RNA was used as a template for RT-PCR. RT-PCR for detection of RET/PTC8 and ACBD5-RET was performed in a 25 μL volume of solution containing 1 × PCR buffer; 200 μM each of dATP, dCTP, dGTP, and dTTP; MgSO4 (3.5 mM for RET/PTC8 and 3.0 mM for ACBD5-RET); 0.4 μM of each primer (PTC8S1, RET-Ex12A4 for RET/PTC8; ACBD5S1, RET-Ex12A4 for ACBD5-RET); and 0.5 U of Platinum Taq DNA polymerase High Fidelity (Invitrogen, Carlsbad, CA). PCR conditions consisted of initial denaturation (95°C for 3 minutes), followed by 40 cycles (denaturation at 95°C for 30 seconds, annealing at 58°C for 30 seconds, and extension at 68°C for 45 seconds), and final extension at 68°C for 5 minutes. To detect expression of BCR, the TK domain of the RET gene, RET/PTC1, and RET/PTC3, RT-PCR was conducted using Platinum Taq DNA polymerase (Invitrogen) in the presence of 1 × PCR buffer; 200 μM each of dATP, dCTP, dGTP, and dTTP; 2.5 mM MgCl2 (2.0 mM for RET/PTC3); and 0.4 μM of each primer at 40 cycles for BCR, RET/PTC1, and RET/PTC3, and 36 cycles for the TK domain. Reaction conditions were as described in RET/PTC8, except for extension temperature (72°C instead of 68°C). Primer sets and annealing temperatures are summarized in Table 1. RT-PCR products of RET/PTC8 and ACBD5-RET were confirmed to be actual products by sequencing after cloning of corresponding bands into a cloning vector.
RT-PCR products other than RET/PTC8 and ACBD5-RET were confirmed to be actual products by digestion of restriction enzymes, BamHI for RET/PTC1 and RET/PTC3, AluI for BCR (TaKaRa), and HaeIII (New England Biolabs, Beverly, MA) for the RET gene TK domain, which existed within each amplified target fragment. For positive control of RT-PCR on the BCR gene, the RET gene TK domain and RET/PTC1, cDNA derived from a human thyroid cell line (TPC1) with RET/PTC1 rearrangement was used as a template. For positive control of RT-PCR on RET/PTC8 and ACBD5-RET, a mixture of 77 base-synthesized nucleotides (PTC8-oligo77 for RET/PTC8 and ACBD5-RET-oligo77 for ACBD5-RET, respectively) and genomic DNA at a molar ratio of 1:1 was used as a template. For negative control, H2O was used as a template. No amplification of RNA without RT was observed.
Results
Concentration of SMART adaptor
All cDNA syntheses in this study were performed with random primer (9 mer), since RNA extracted from archival formalin-fixed, paraffin-embedded thyroid cancer tissue specimens was invariably degraded to some degree. Using RNA prepared from each of two in-house controls harboring RET/PTC1 and RET/PTC3 rearrangements, first we examined the effects of concentration of SMART adaptor on the amplification efficiency by SMART RACE-PCR with gene-specific and adaptor-specific primers. One microliter of various concentrations of SMART adaptor was added to the reaction mixture immediately after cDNA synthesis had been completed. The mixture was further incubated at 42°C for 60 minutes in the presence of SMART adaptor. Since the total length of the SMART adaptor and the 5′ portion of exon 12 of RET is 55 bp, the length of amplified target fragments requires more than 70 bp to identify the counterpart gene. In addition, it is important to conduct cloning of obvious and intense fragments for successful isolation and identification of the counterpart gene. Therefore, among clear-cut fragments of more than 70 bp, fragments with a BamHI digestion site were determined as target candidates, since the fragments amplified by SMART RACE-PCR included BamHI recognition site at the 5′ end of exon 12 of the RET gene. All fragments in Figure 1A–D that could be recognized as a band were confirmed to be digested with BamHI. As shown in lanes 2–4 of Figure 1A and C, PCR products looked like smears without clear and intense bands when adaptor concentration was high (2–10 μM). PCR products with 0.2–1 μM of SMART adaptor revealed an obvious and intense band of more than 70 bp compared with the bands at other higher concentrations (lanes 5–7, Fig. 1A and C, indicated by asterisks), though the sizes of these target candidate fragments differed somewhat among various concentrations of SMART adaptor. Treatment with 0.1 μM of SMART adaptor showed that amplified bands of more than 70 bp in length were observed only infrequently (lane 8, Fig. 1A, C).
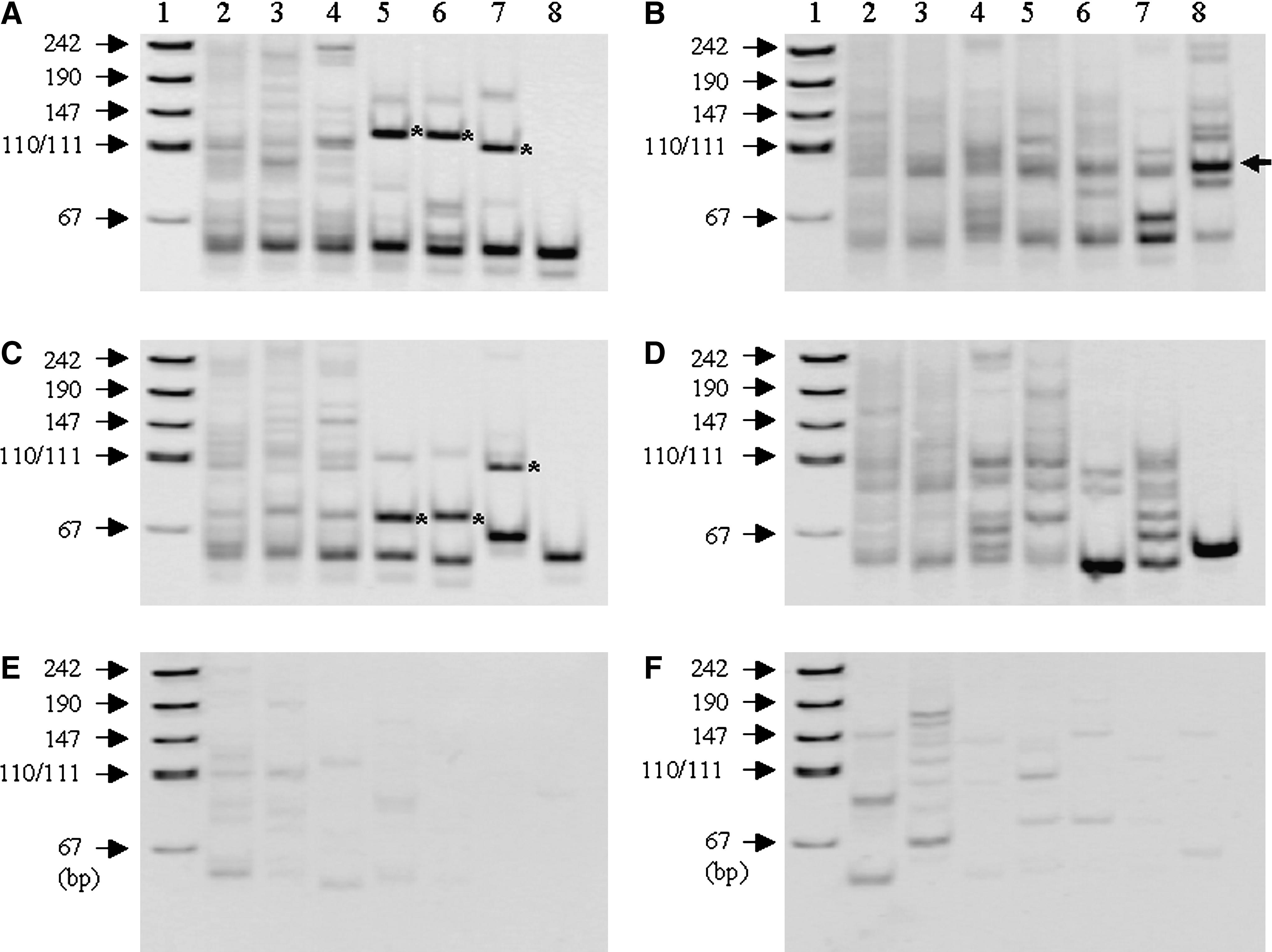
Effects of concentration and addition timing of SMART adaptor on improved SMART RACE-PCR amplification. (
Addition timing of SMART adaptor
When cDNA was synthesized in the presence of SMART adaptor (2–10 μM), electrophoretic images of PCR products were similar to those in the case of addition of adaptor after completion of the cDNA synthesis (compare lanes 2–4 in Fig. 1B and D with lanes 2–4 in Fig. 1A and C). When cDNA was prepared in the presence of a lower concentration (0.2–1 μM) of SMART adaptor, the electrophoretic patterns differed from those where adaptor was added after completion of cDNA synthesis (compare lanes 5–7 in Fig. 1B and D with lanes 5–7 in Fig. 1A and C, respectively). Namely, weak bands of more than 70 bp were observed (lanes 5–7, Fig. 1B, D). Although treatment with 0.1 μM of SMART adaptor occasionally produced clear and intense bands (lane 8, Fig. 1B, indicated by an arrow), amplified bands of more than 70 bp in length were rarely observed (lane 8, Fig. 1D).
When RNA extracted from archival samples with no detectable expression of RET gene TK domain was used, several weak bands were observed regardless of the addition timing of SMART adaptor. However, few bands (more than 70 bp) were found in the cases of 0.1, 0.2, or 0.4 μM of SMART adaptor (lanes 6–8, Fig. 1E, F). Not all weak bands in Figure 1E and F could be digested with BamHI. No bands were observed when H2O was used as template for RACE-PCR. In addition, SMART RACE-PCR without reverse transcription produced no amplified bands whatsoever.
Incubation time
We examined the effects of incubation time after addition of adaptor on amplification by SMART RACE-PCR using RNA from specimens with RET/PTC1. After SMART adaptor was added to cDNA solution to give a final concentration of 0.2 μM, the reaction was incubated for various durations at 42°C. Among target candidate bands of about 90 bp (lanes 2–6, Fig. 2, indicated by asterisks) found in SMART RACE-PCR products until 45 minutes of incubation time, a case treated for 45 minutes revealed the most intense band (lane 6, Fig. 2). Incubation for 60 minutes resulted in a larger intense target candidate band (lane 7, Fig. 2) than did other incubation times. In the case of 90-minute incubation (lane 8, Fig. 2), the larger candidate band disappeared and a weak 90 bp candidate band reappeared (indicated by an asterisk).
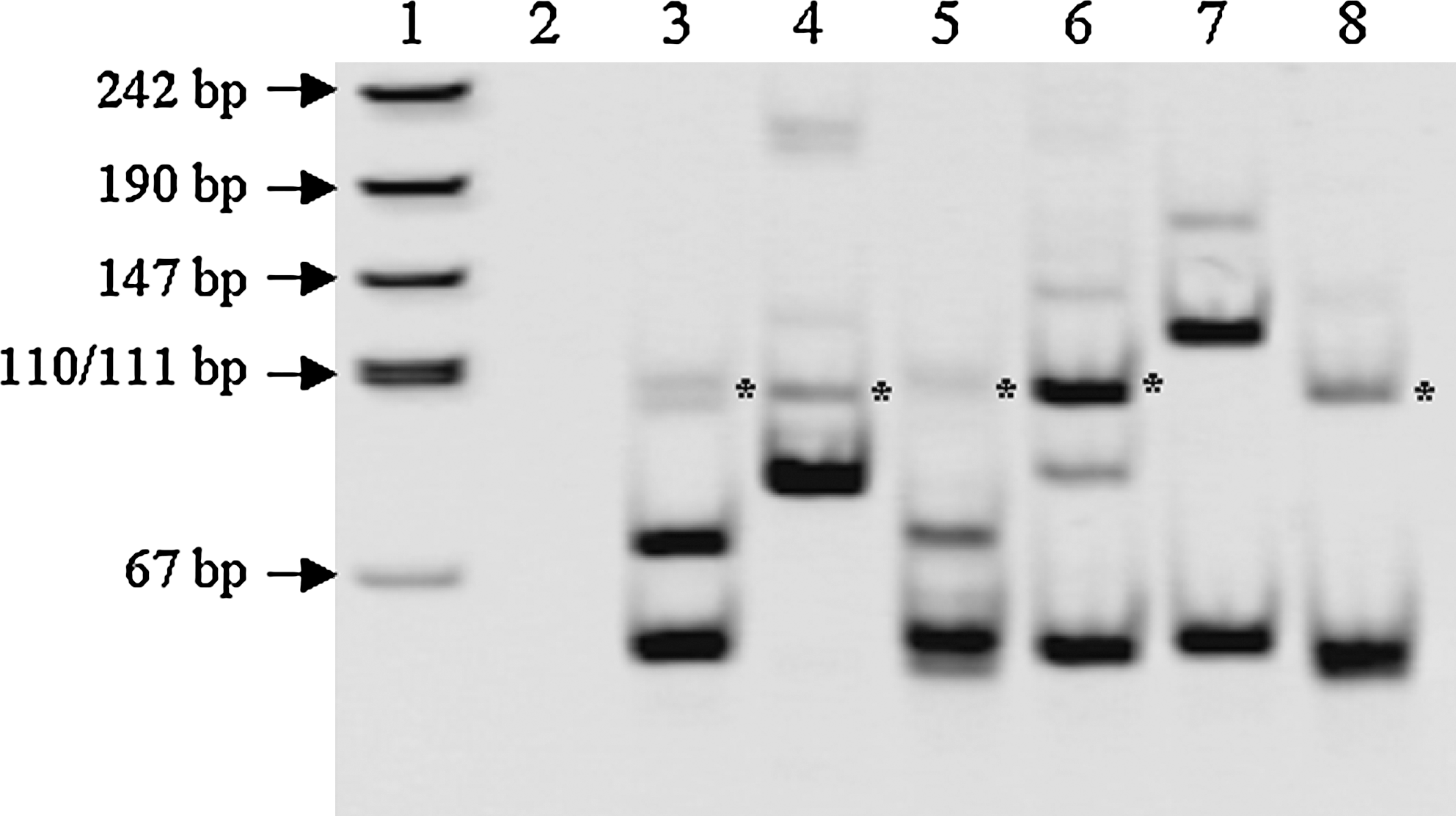
Effect of incubation time on improved SMART RACE-PCR amplification. The reaction was incubated for various times at 37°C after SMART adaptor was added to cDNA solution to give a final concentration of 0.05 μM (lane 2, 0 minute; lane 3, 10 minutes; lane 4, 20 minutes; lane 5, 30 minutes; lane 6, 45 minutes; lane 7, 60 minutes; lane 8, 90 minutes). Lane 1 indicates pUC19-MspI digest for DNA size marker. The asterisks indicate fragments of about 90 bp with BamHI recognition site.
We next examined how efficiently cDNA fragments of the target gene can be isolated by this improved SMART RACE method. Target candidate bands of about 100 bp (lane 7, Fig. 1A) and 90 bp (lane 7, Fig. 1C) in the second PCR products were eluted from 8% acrylamide gel and cloned onto a pUC118 plasmid vector. Plasmid DNA with a longer insert than 70 bp was sequenced. As shown in Table 2, all 36 screened colonies proved to be plasmid DNA harboring RET/PTC1 fragments of 97 bp in length. RET/PTC3 fragments with a length of 89 bp were also detected in 35 out of 36 colonies, as shown in Table 2. This suggests that almost all candidate fragments amplified by the improved SMART RACE method are actually target cDNA fragments.
Thyroid cancer with RET/PTC1.
Thyroid cancer with RET/PTC3.
One clone failed to perform sequencing.
RET/PTC8 in thyroid cancer tissue from A-bomb survivors
RNA was extracted from unbuffered formalin-fixed, paraffin-embedded papillary thyroid cancer tissue specimens from 64 subjects exposed to A-bomb radiation, and BCR gene expression was detected in 52 of the 64 cases. Among these 52 cases, 11 showed expression of RET gene TK domain, of which 9 cases had RET/PTC1 or RET/PTC3, as shown in Table 3. Two papillary thyroid cancer cases showing no RET/PTC1 or RET/PTC3 rearrangement were examined for type of rearrangement using this improved SMART RACE method. The target band in the second PCR products (Fig. 3A, indicated by an arrow) was eluted and cloned into a cloning vector. Since 23 out of 24 white colonies contained plasmid DNA with BamHI site and a longer insert than about 70 bp, plasmid DNA in the 23 colonies was sequenced. The sequencing of the portion upstream of RET exon 12 found in all clones was identical to the 5′ part of the kinectin 1 gene, whose insert length was 93 bp (Fig. 3B). As shown in Figure 3C, the band corresponding to RET/PTC8 was also detected in the papillary thyroid cancer specimen from the same A-bomb survivor by RT-PCR. Further, the improved SMART RACE method enabled us to identify a novel rearrangement of the RET gene in papillary thyroid cancer of another A-bomb survivor. This new type of RET/PTC, whose partner gene is acyl coenzyme A binding domain 5 (ACBD5) located on chromosome 10p, is now being analyzed for its tumorigenicity. Expression of the ACBD5-RET fusion gene was confirmed in RNA from the cancer specimens by RT-PCR (Fig. 3C).
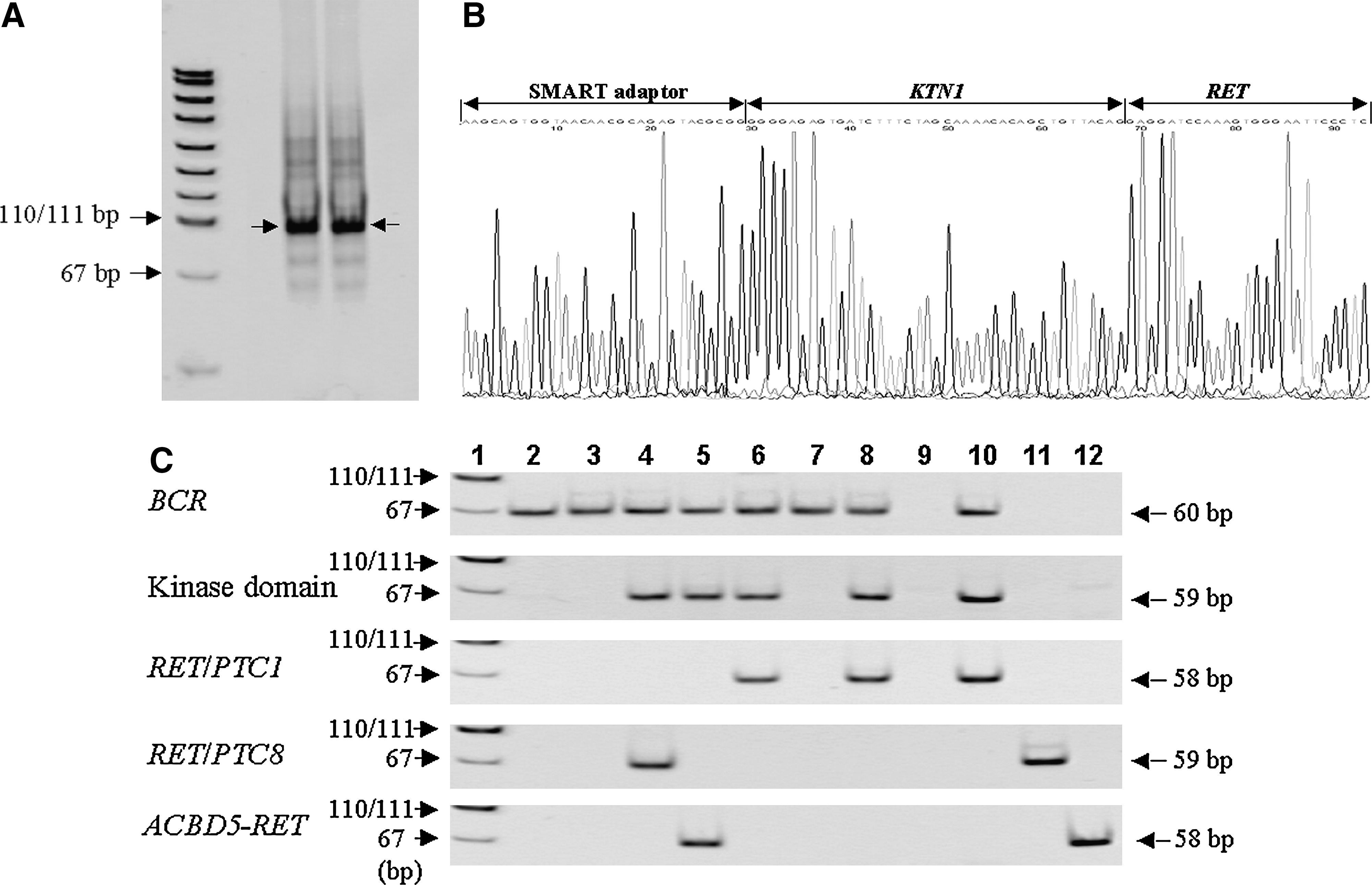
Identification of RET/PTC8 in papillary thyroid cancer from one A-bomb survivor. (
One case carried both RET/PTC1 and RET/PTC3.
TK, tyrosine kinase.
Discussion
RACE reaction with RNA extracted from fresh tissues or cells can be easily and successfully performed using the SMART RACE kit (Clontech). On the other hand, as far as we know, there are thus far no reports on RACE with RNA extracted from archival unbuffered formalin-fixed, paraffin-embedded tissue samples. We succeeded in identification and isolation of a partner gene of rearranged RET using RNA extracted from archival unbuffered formalin-fixed and paraffin-embedded thyroid cancer tissue specimens by improved SMART RACE method. A series of experiments from cDNA synthesis to PCR was conducted at least three times. Similar electrophoresis patterns could be reproduced, though the sizes of the target bands differed somewhat. Size differences of amplified fragments between experiments may largely have been due to degradation of RNA extracted from archival samples, since such a phenomenon was not observed when cDNA was prepared with the random primer (9 mer) using intact RNA from fresh samples (data not shown).
We found that the SMART RACE method enabled isolation of 5′ upstream fragments in target cDNA with RNA extracted from archival formalin-fixed, paraffin-embedded tissue specimens by adjusting the concentration and the timing of the addition of SMART adaptor. The optimal concentration of SMART adaptor (ultimately around 0.2 μM) was lower than that in conventional SMART RACE with oligo (dT) using intact RNA (9). This finding was contrary to our expectation that a higher concentration of adaptor would be required because of degradation of the RNA extracted from archival formalin-fixed and paraffin-embedded tissue. Since a large amount of SMART adaptor brought in by cDNA used as a PCR template has a GGG sequence at the 3′ end, this adaptor may cause nonspecific amplification by the first PCR. As a result, specific and efficient RT-PCR may be hindered. Further, when cDNA in the presence of SMART adaptor was prepared with random primers (9 mer) using RNA extracted from archival tissue specimens, we could not obtain the clear and intense target candidate bands observed in the case of addition of adaptor after completion of cDNA synthesis (Fig. 1A and C vs. 1B and D). However, when cDNA was prepared with random primers using intact total RNA, target candidate bands could be detected regardless of the timing of adding SMART adaptor (data not shown). This suggests that when degraded RNA was used, a larger amount of short cDNA primed with SMART adaptor may be produced, possibly interfering with specific PCR amplification.
In addition to the concentration and the timing of the addition of SMART adaptor, length of incubation time was also an important factor for efficient amplification by our SMART RACE-PCR: optimal incubation time was relatively long (45–60 minutes) despite addition of adaptor after cDNA synthesis completion. Thus, although amplification efficiency by SMART RACE-PCR largely depended on concentration of SMART adaptor, timing of adaptor addition, and incubation time when cDNA was prepared with random primer using RNA extracted from archival formalin-fixed and paraffin-embedded tissue specimens, unlike SMART RACE with intact RNA, we found that the improved SMART RACE method achieved successful isolation of 5′ upstream fragments in target cDNA with RNA extracted from archival unbuffered formalin-fixed, paraffin-embedded tissue specimens.
A rare rearrangement of the RET gene, RET/PTC8, previously identified in papillary thyroid cancers in children from areas contaminated by the Chernobyl accident (12), was detected in papillary thyroid cancer of one A-bomb survivor exposed to more than 2 Gy of radiation. Further, a novel rearrangement of the RET gene (ACBD5-RET fusion gene) was identified in papillary thyroid cancer of another A-bomb survivor exposed to more than 1.5 Gy. It is interesting to note that a rare RET/PTC8 rearrangement and a novel ACBD5-RET fusion gene were identified in papillary thyroid cancers in A-bomb survivors exposed to a relatively high radiation dose.
Since this improved SMART RACE method achieves effective amplification of the unknown 5′ upstream region of certain genes with even 10 ng of total RNA extracted from archival formalin-fixed and paraffin-embedded tissue specimens, the method is expected to prove useful in molecular analyses using archival tissue samples of limited quantity.
Footnotes
Acknowledgments
The RERF, Hiroshima and Nagasaki, Japan, is a private, nonprofit foundation funded by Japan's Ministry of Health, Labour, and Welfare and the U.S. Department of Energy, the latter in part through the National Academy of Sciences. This publication was supported by RERF Research Protocols RP 5–02 and B31-03, and in part by a Grant-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science, and Technology, and a Grant-in-Aid for Cancer Research from Japan's Ministry of Health, Labour, and Welfare.
Disclosure Statement
The authors declare that they have no commercial associations that might create a conflict of interest in connection with this article.