
Abstract
Background:
The degradation of many nuclear receptors is controlled by ligand-binding and mediated by the ubiquitin-proteasome pathway. However, the mechanisms implicated in thyroid hormone receptor (TR) degradation remain unclear. Our objective was to define the kinetics, mechanisms, and sub-cellular fractions involved in TRs degradation.
Methods:
We used pulse-chase analyses, time-course experiments carried out in presence of cycloheximide (to inhibit new protein synthesis), and biochemical fractionation with Western blot analyses to determine the kinetics of the degradation of the TRβ isoform, TRβ1, in transiently transfected QBI-HEK 293A cells.
Results:
We observed that TRβ1 degradation is mediated by the proteasome pathway. Also, the kinetics of TRβ1 degradation is atypical due to the co-existence of more than one TRβ1 population, located in different cellular compartments and having different stability profiles. Moreover, TRβ1 degradation was unaffected by a mutation in its putative PEST motif, which confers turnover of other proteins.
Conclusion:
Our findings introduce novel evidence suggesting that stable and unstable forms of TRβ1, which might have distinct functions, co-exist in cells.
Introduction
Even before the cloning and identification of TRs isoforms, it was reported that THs influence the steady-state of their NRs in the rat pituitary GH1 cell line (11,12). First, it was observed that T3 treatment reduces the nuclear binding capacity of L-125I-T3 by a time- and dose-dependent depletion of the NRs (11). Then, using dense amino acid labeling, Raaka and Samuels (12) demonstrated that T3 decreases the steady-state of its specific NRs by reducing their synthesis and half-life. Recently, reports showed that TRs are ubiquitinated in the absence and presence of T3 and that ligand-induced decrease of TR levels seems to be mediated by the ubiquitin-proteasome pathway (13,14). However, we cannot, from these investigations, conclude if only T3 affects the degradation of TRs or if other mechanisms are also implicated.
Thus, mechanisms implicated in TR decay remain unclear. Therefore, the aim of this study was to determine the kinetics of TRβ1 degradation. This question is relevant since TR degradation influences overall TH action. For this purpose, we used an influenza hemagglutinin (HA) epitope-tagged TRβ1 transiently transfected in QBI-HEK 293A cells to perform pulse-chase analyses that are the gold standard to study protein degradation. We also used time-course experiments carried out in presence of cycloheximide and small-scale biochemical fractionation with Western blot analyses to better understand the mechanisms involved in TRβ1 degradation.
Materials and Methods
Plasmid constructions
The expression vector pcDNA5/FRT/HA-TRβ1 was prepared by polymerase chain reaction (PCR) using the forward primer 5′-CCCAAGCTTGGGGCCGCCGCCATGTACCCATACGACGTGCCAGACTACGCTCTCGAGACTCCCAACAGTATGACAGAAAATGGCCTTACAGCC-3′, the reverse primer 5′-ATAGTTTAGCGGCCGCATTCTTATCTAATCCTCGAACACTTCCAAGAACAA-3′ and hTRβ1 (accession number NM_000461) as a template. The PCR product, where the HA tag was at the N-terminus of TRβ1, was introduced in the HindIII and NotI restriction sites of pcDNA5/FRT (Invitrogen). The empty vector pcDNA5/FRT/HA was obtained by digestion with XhoI. The sequence was confirmed by restriction enzyme analysis and DNA sequencing (McGill University and Genome Québec Innovation Center).
A point mutation in the putative PEST motif [a peptide sequence that is rich in proline (P), glutamic acid (E), serine (S), and threonine (T)] of TRβ1 was generated by PCR mutagenesis using Phusion Site-Directed Mutagenesis Kit (New England BioLabs) and the forward 5′-TGCAGAAGTCCATCGGGCACAAGCCAGAGCCCGCCGACGAGGAATGGGAG-3′ and reverse 5′-GCTCTTCCCGCCGTCTTTTCTCCCGGTTCT-3′ primers. The resulting HA-TRβ1/T215A mutant was verified by sequencing.
Cell culture
QB1-HEK 293A cells (Q-BIOgene), a cell line derived from human embryonic kidney cells, were grown in Dulbecco's modified Eagle's medium (DMEM; Wisent) supplemented with 10% fetal bovine serum (FBS; Wisent), at 37°C in a humidified 5% CO2 atmosphere.
Transient transfections
Sixteen hours before transfection, cells were split and seeded in 60-mm dishes. Cells were transiently transfected using Lipofectamine 2000 (Invitrogen), according to the protocol supplied by the manufacturer. Five hours after transfection, cells were pooled, split, and reseeded in 60-mm dishes, such that the amount and distribution of HA-TRβ1 are the same in all dishes, thus preventing variations in transfection efficiency. Cells were analyzed 24 hours after transfection.
Pulse-chase experiments
Pulse-chase experiments were performed as described by Tansey (15,16). Briefly, cells were washed twice with phosphate-buffered saline (PBS), and starved of methionine, cysteine, and THs by an incubation of 30 minutes at 37°C in methionine/cysteine-free DMEM (Wisent), supplemented with 5% dialyzed FBS (Wisent) stripped of hormones by anion-exchange resin and charcoal. Cellular proteins were pulse-labeled by incubating each 60-mm dish of cells with 500 μCi of [ 35 S]-methionine/cysteine ( 35 S-EasyTag Express Protein Labeling Mix; Perkin Elmer) for 1 hour at 37°C. The radioactive medium was then removed, and cells were washed with PBS and re-fed with DMEM containing 2 mM methionine, 2 mM cysteine (Wisent), 5% dialyzed FBS stripped of hormones, and 100 nM T3 or the vehicle alone. The T3 concentration was chosen according to previous TRs degradation and distribution studies (13,17,18). Cells were incubated for the indicated chase times before being collected and frozen in liquid nitrogen. Labeled proteins were recovered by denaturing immunoprecipitation (IP). Where indicated, the proteasome inhibitor MG132 (Sigma) at a final concentration of 50 μM or the vehicle (DMSO) was applied to cells during amino acid starvation and renewed during the chase.
Denaturing IPs
Denaturing IPs were performed as described by Tansey (19) with minor modifications. Pre-IP aliquots were sampled before the addition of proteins A/G-agarose as an input control. Total labeled HA-tagged TRβ1 proteins were precipitated using 4 μL of anti-HA antibody (MMS-101P; Covance). IP, along with pre-IP aliquots, were loaded in duplicate and detected using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and autoradiography. HA-TRβ1 levels and total labeled proteins were quantified (in CPM) with Instant Imager (Packard) and the mean of a duplicate was used for analysis. The experimental conditions of denaturing IP were optimized to ensure that no artefacts were introduced by limiting amounts of reagents.
Calculation of the kinetics of TRβ1 degradation
At each chase time, HA-TRβ1 levels (the value of IP) were divided by total labeled proteins (the value of the pre-IP) and expressed as a percentage of the initial level. Results (the mean ± SEM of three independent experiments) were approximated by exponential function according to the best coefficient of determination (plateau followed by one phase decay, in the absence of T3 and one phase decay, in the presence of T3) using GraphPad Prism software. Receptor half-life was calculated with the following equations:
where y indicates the relative quantity of HA-TRβ1 (%); x, the chase time (minute); x0, the time at which the decay begins (minute); and y0, the y value when x is 0 (the initial percentage of HA-TRβ1 protein was constrained to 100); the plateau refers to the y value at infinite time (% of HA-TRβ1); k is the rate constant (expressed in inverse min). From these equations, the half-life of HA-TRβ1 corresponds to the x value when y is 50.
Time-course studies carried out in the presence of cycloheximide
Time-course studies carried out in the presence of cycloheximide were conducted in conditions similar to pulse-chase experiments. Cells were starved of THs by incubation for 90 minutes at 37°C in DMEM supplemented with 10% hormone-stripped FBS. Cells were re-fed with DMEM supplemented with 60 μg/mL of cycloheximide (Sigma), 10% hormone-stripped FBS, and 100 nM T3 or the vehicle alone and incubated for the indicated chase times. Where indicated, MG132 (50 μM) or the vehicle (DMSO) was applied to cells 30 minutes before the end of TH starvation and renewed during the chase. Cells were collected and frozen in liquid nitrogen. Isolation of proteins and Western blot analyses were performed as described below.
Isolation of proteins
Small-scale biochemical fractionation was modified from that of Wysocka et al. (20). Briefly, cells (2.5 × 106) were collected, washed in PBS, quickly frozen in liquid nitrogen, resuspended in 200 μL of buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol, 0.12% Triton X-100, and protease inhibitor cocktail [Roche]) and incubated on ice for 20 minutes. Nuclei (fraction P1) were collected by centrifugation (5 minutes, 1300 g). The supernatant (fraction S1) was clarified by high-speed centrifugation (5 minutes, 20,000 g) and the supernatant (cytoplasmic fraction S2) was collected. The integrity of the P1 nuclei fraction was verified using microscopy before being washed in buffer A and lysed for 30 minutes in 50 μL of buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and protease inhibitor cocktail). Insoluble chromatin (fraction P3) and soluble nuclear fractions (fraction S3) were separated by centrifugation (5 minutes, 1700 g). The P3 fraction was washed and resuspended in 50 μL of buffer B and sonicated.
Whole-cell protein extracts were performed in denaturing conditions as described by Battista et al. (21) and modified as follows: the cell pellets were incubated in boiling buffer (2% SDS, 1% Triton X-100 in PBS), heated at 95°C for 10 minutes, and sonicated.
Western blot analyses
All samples were assayed for protein content before Western blotting. Proteins were resolved by electrophoresis on 10 or 15% SDS-PAGE and transferred onto polyvinyl difluoride membrane (Roche). Proteins were detected by Western blot using anti-HA (MMS-101P; Covance), anti-proliferating cell nuclear antigen (PCNA) (P8825; Sigma), anti-IκBα (610690; BD Transduction Laboratories), anti-Histone H2B (07-371; Millipore), anti-Lamin B (sc-6217; Santa Cruz Biotechnology), and anti-GAPDH (ab9485; Abcam) antibodies. When it was required, membrane was stripped (twice for 15 minutes in 0.2 N NaOH and twice for 15 minutes in PBS), blocked, and reblotted for a maximum of 3 times. Quantification of band intensity was determined using ImageJ software.
Results
TRβ1 decay is mediated by the proteasome pathway and is atypical
TRβ1 levels in QB1-HEK 293A cells are too low to allow the study of TRβ1 stability by pulse-chase analyses (data not shown). Thus, we used an HA-tagged TRβ1 (with a transcriptional activity identical to that of the untagged TRβ1, as evaluated by luciferase assays [data not shown]) transiently transfected in QB1-HEK 293A cells to investigate the degradation of TRβ1 (Fig. 1). The addition of the proteasome inhibitor MG132 completely blocked the proteolysis of TRβ1 both in the absence and in the presence of T3, confirming that TRβ1 is degraded by the proteasome pathway (Fig. 1a, b). T3 treatment seems to alter the pattern of TRβ1 degradation toward more rapid degradation (Fig. 1a), but the difference between half-life (estimated at 134 minutes in the absence of T3 vs. 103 minutes in the presence of T3) was not statistically significant (Fig. 1c).
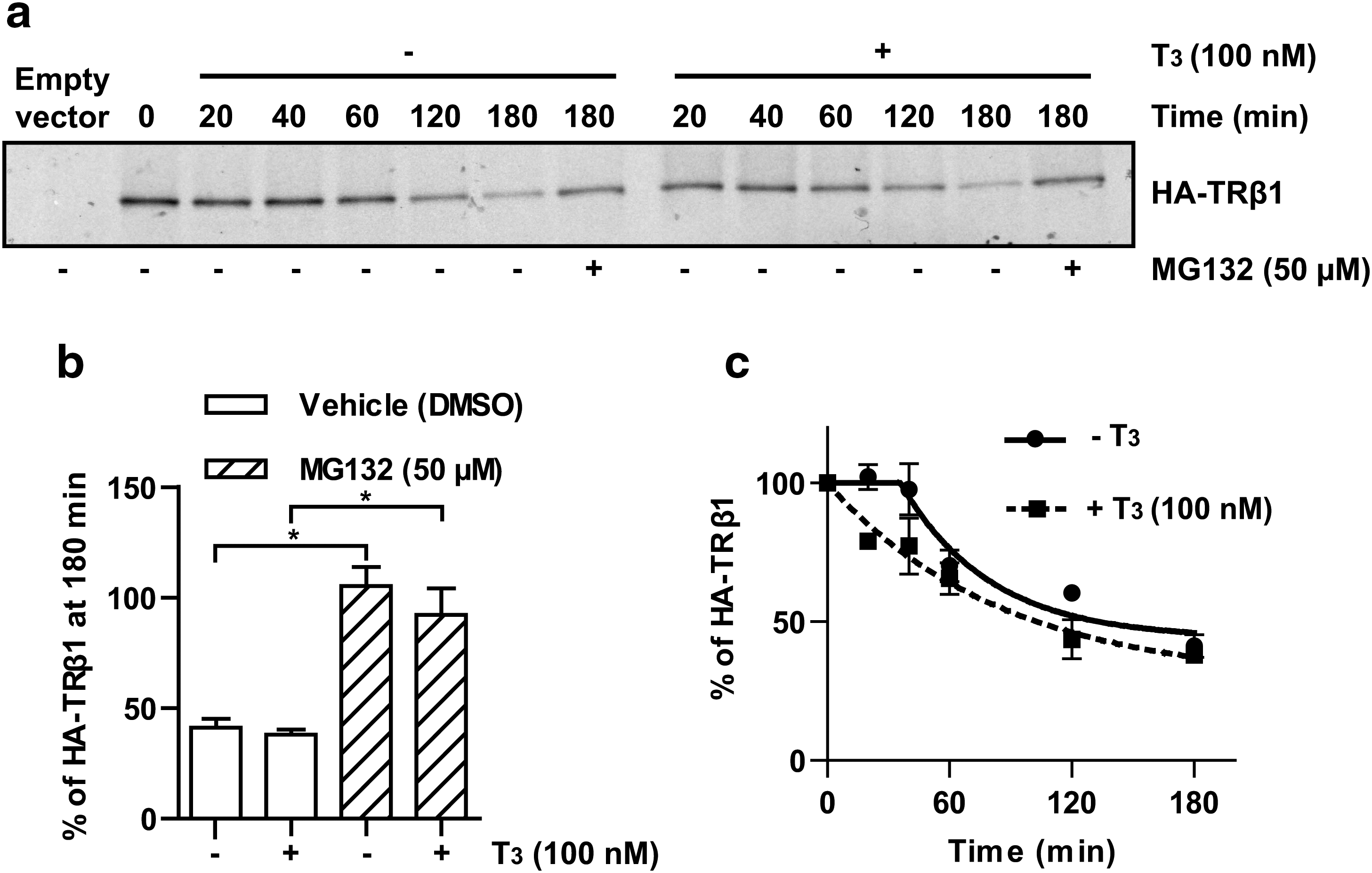
Study of TRβ1 decay by pulse-chase experiments. QB1-HEK 293A cells were transiently transfected with pcDNA5/FRT/HA-TRβ1 and subjected to pulse-chase analyses in the absence or presence of 100 nM of T3. Total labeled HA-TRβ1 proteins were recovered by denaturing immunoprecipitation with the anti-HA antibody, analyzed by SDS-PAGE, and observed by autoradiography. (
Further, the degradation pattern of TRβ1 is atypical. Indeed, in the absence of T3, the degradation of TRβ1 started only 30 to 40 minutes after the beginning of the chase, whereas in the presence of T3, the degradation began immediately (Fig. 1c). Both in the absence and in the presence of T3, the rate of TRβ1 degradation slowed down around 120 minutes of chase and TRβ1 levels reached a plateau. Remaining TRβ1 levels were higher in the absence of T3 than in the presence of T3: the residual TRβ1 levels, extrapolated from Eqs. (1) and (2), were 44% in the absence of T3 versus 28% in the presence of T3. Similar kinetics of TRβ1 degradation were also observed using time-course studies carried out in the presence of cycloheximide followed by detection of TRβ1 by Western blot analyses, suggesting that this pattern of degradation is not caused by technical artefacts (data not shown). Thus, our data suggest that TRβ1 in cells are not all degraded at the same rate.
TRβ1 prevails in the nuclear fraction containing chromatin and nuclear matrix components
We thus hypothesized that more than one population of TRβ1 co-exist in cells. To verify this hypothesis, QB1-HEK 293A cells transiently transfected with HA-TRβ1 were fractionated into cytosolic fraction (S2), soluble nuclear fraction (S3), and insoluble nuclear fraction containing chromatin and nuclear matrix components (P3). HA-TRβ1 was detected by Western blot analyses (Fig. 2). TRβ1 was detected in all three fractions, but its expression was predominant (67.6%) in the P3 fraction. Appropriate fractionation was verified by Western blot analyses of well-documented cellular proteins: IκBα, which localizes to the cytoplasm, histone H2B, a component of chromatin, Lamin B, a component of nuclear matrix (22), and PCNA, a nuclear protein (23). As expected, IκBα was present exclusively in the S2 fraction, whereas histone H2B and Lamin B resided only in the P3 fraction. PCNA was detected in S3 but also in the S2 fraction, even though the lysis of cytoplasmic membrane and the integrity of nuclear envelope were verified with microscopic analysis (data not shown). One possible explanation is that S2 fraction may contain small amounts of soluble nuclear components owing to permeabilization of the nuclear membrane by nonionic detergents, as suggested by Wysocka et al. (20).
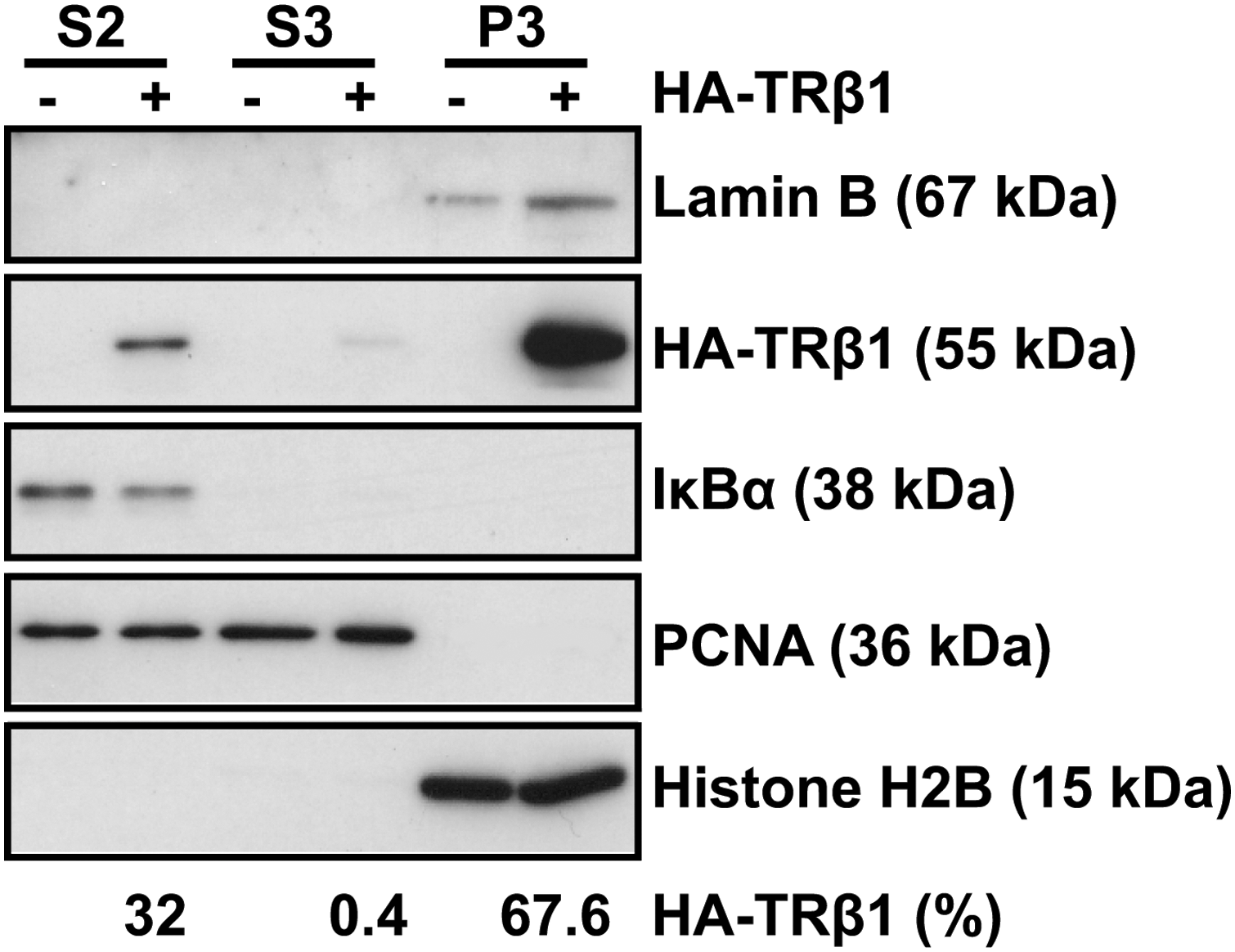
TRβ1 is found mainly in the chromatin and nuclear matrix cellular fraction. QB1-HEK 293A cells were transiently transfected with pcDNA5/FRT/HA-TRβ1 (+) or empty vector (−) and separated into cytosolic fraction (S2), soluble nuclear fraction (S3), and insoluble nuclear fraction containing chromatin and nuclear matrix components (P3). Proteins (20 μg) were separated on 15% SDS-PAGE and revealed by immunoblotting (IB) using antibodies against HA, PCNA, H2B, IκBα, and Lamin B. HA-TRβ1 levels were determined by densitometric analysis and normalized with respect to the migrated fraction. Results are expressed as a percentage of the total amount of HA-TRβ1 in the three fractions. PCNA, proliferating cell nuclear antigen.
Stable and unstable forms of TRβ1 co-exist in cells
We then analyzed TRβ1 decay in the three cellular fractions with time-course studies carried out in the presence of cycloheximide to inhibit new protein synthesis (Fig. 3). The use of the proteasome inhibitor prevented TRβ1 degradation in all fractions (Fig. 3a). The kinetics of TRβ1 disappearance in each fraction was very different. In the S2 cytosolic fraction, TRβ1 was almost stable for 60 minutes in the absence of T3, whereas TRβ1 levels quickly decreased in the presence of T3 (Fig. 3a, b). In the S3 soluble nuclear fraction, TRβ1 disappeared more rapidly in the presence of T3 than in the absence of T3 (Fig. 3a, c). In the P3 chromatin and nuclear matrix fraction, TRβ1 was stable in the absence of T3 (Fig. 3a, d). However, after 20 minutes of T3 treatment, TRβ1 levels increased in the P3 fraction and returned to the initial level after 40 minutes (Fig. 3d), suggesting a rapid T3-induced movement of TRβ1 from cytosolic (S2) and/or soluble nuclear (S3) populations toward the chromatin and/or nuclear matrix. After that, TRβ1 levels decreased by 30% between 60 and 120 minutes in the P3 fraction. These results indicate that stable and unstable forms of TRβ1 co-exist in cells.
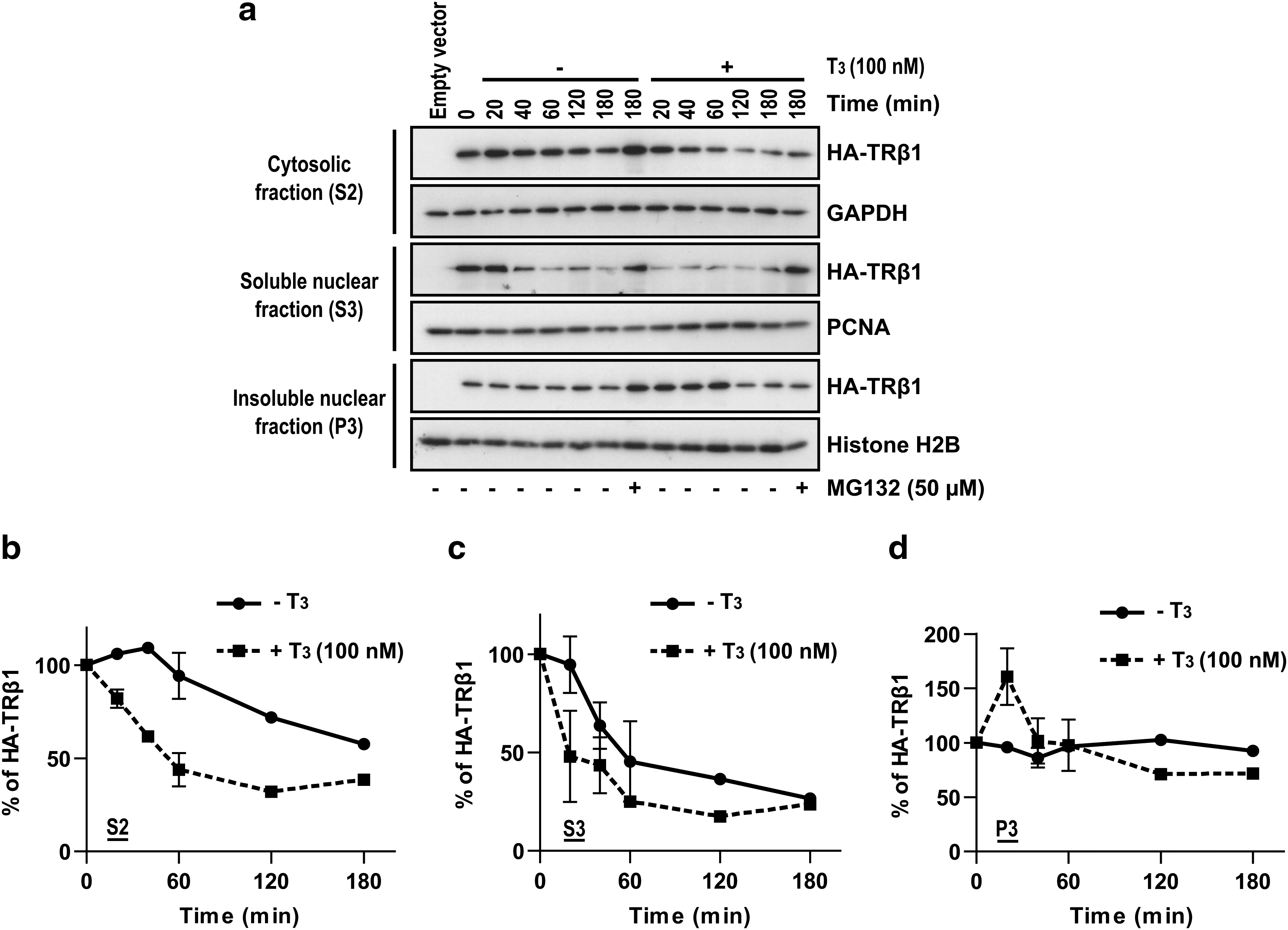
Stable and unstable forms of TRβ1 co-exist in cells. QB1-HEK 293A cells were transiently transfected with pcDNA5/FRT/HA-TRβ1 and treated for the indicated length of time with cycloheximide (60 μg/mL) to inhibit protein synthesis, with or without T3 (100 nM) and with or without MG132 (50 μM), and then the biochemical fractionation was performed. Proteins (30 μg of S2, 20 μg of S3, and 20 μg of P3) were separated on 15% SDS-PAGE. HA-TRβ1 was detected by IB with anti-HA antibody. GAPDH was used as loading control for S2, PCNA for S3, and H2B for P3. (
A putative PEST motif is not involved in TRβ1 degradation
Many proteins contain PEST motifs, a sequence rich in proline, glutamic acid, serine, and threonine residues, that are involved in the recognition and degradation of target proteins by the ubiquitin-proteasome pathway (3,24). Phosphorylation of threonine and serine residues could contribute to the rapid degradation of proteins that contain PEST motifs (24). PESTfind software (

A putative PEST motif is not involved in TRβ1 degradation. QB1-HEK 293A cells were transiently transfected with pcDNA5/FRT/HA-TRβ1/T215A. Twenty-four hours after transfection, cells were treated for the indicated length of time with cycloheximide (60 μg/mL), with or without T3 (100 nM) and with or without MG132 (50 μM), and then whole-cell protein extracts were performed. HA-TRβ1/T215A was detected by IB using the anti-HA antibody. PCNA was used as a loading control. (
Discussion
In this study, we characterized the kinetics of TRβ1 degradation using pulse-chase analyses, time-course experiments carried out in presence of cycloheximide, and biochemical fractionation with Western blot analyses. Our results confirm that TRβ1 degradation is mediated by the proteasome pathway (Fig. 1b). We also provide evidence that TRβ1 decay is atypical due to the co-existence of different compartment-specific TRβ1 populations having different kinetics of degradation (Fig. 3). Biochemical fractionation of TRβ1 showed that TRβ1 is present in the cytoplasm and predominant in the nucleus of cells (Fig. 2), which is consistent with confocal microscopy studies (17,18). The majority of TRβ1 is localized in the insoluble nuclear fraction containing chromatin and nuclear matrix components (P3, Fig. 2). Moreover, our results suggest that the putative PEST motif of TRβ1 is not required for its degradation by the proteasome (Fig. 4). These results confirm previous reports that demonstrated that T3 treatment creates an imbalance in the steady-state of TRs (11,12) and that degradation of TRs is mediated by the proteasome pathway (13,14), and reveal several new observations that provide a more complete understanding about the mechanisms involved in TRβ1 degradation. However, we acknowledge that the use of overexpressed TRβ1 is a potential limit of our study and restricts conclusions to this isoform.
We estimated the half-life of TRβ1 at 134 minutes and 103 minutes in the absence and the presence of T3, respectively (Fig. 1c). Apparent discrepancies between these values and those previously reported may be explained. Raaka and Samuels (12) studied all TR isoforms present in the nucleus of the rat pituitary GH1 cell line as an homogeneous population, whereas our values are for the human TRβ1 isoform alone in whole cells. Also, Dace et al. (13) used time-course experiments and Western blot analyses without protein synthesis inhibitor; therefore, the effect of T3 on TR levels that they observed may be due to an increase in TR degradation but also to variations in TR synthesis. Our study, using gold-standard pulse-chase analyses, thus is probably the more precise available that describes the TRβ1 half-life.
Biochemical fractionation followed by Western blot analyses allowed us to study separately TRβ1 subcellular populations, whereas pulse-chase experiments, examining total TRβ1, do not allow the detection of compartment-specific events. Results of TRβ1 degradation in subcellular compartments (Fig. 3) provide evidence that the atypical decay observed in pulse-chase experiments (Fig. 1) results from the co-existence of distinct populations of TRβ1 with different stabilities. Our results also indicate that certain TRβ1 populations are rapidly degraded, whereas others seem to be protected from proteasomal degradation (Fig. 3). The significance of this finding and how these different populations are regulated remain unknown. The co-existence of metabolically stable and unstable forms of a protein was also observed with another transcription factor, the oncoprotein Myc (25). A possible explanation for the stable forms of TRβ1 in the cytoplasm (Fig. 3b, in the absence of T3) and in the insoluble nuclear fraction (Fig. 3d, in the absence and presence of T3) could be their protection from degradation by post-translational modifications, such as phosphorylation, that stabilize TRβ1 (26,27). Further, TRβ1 levels in the insoluble nuclear fraction (Fig. 3d) might be modified by TRβ1 interaction with chromatin. This is suggested since histone acetylation, possibly coupled with T3 action, changes the affinity of the receptor for the chromatin, which may affect TR levels (28,29). Our studies thus open novel research areas and a better characterization of each population is required.
We observed different kinetics of TRβ1 disappearance depending on the cellular fraction (Fig. 3). Confocal microscopy studies demonstrated that T3 induces a rapid cytoplasm-to-nucleus translocation (18) and intranuclear reorganization of TRβ1 (17). Therefore, in addition to TRβ1 degradation by the proteasome, a movement of TRβ1 from cytosolic (S2) and/or soluble nuclear (S3) populations toward the chromatin and/or nuclear matrix (P3) probably partly explains the T3-induced decrease in TRβ1 levels in the cytoplasm (S2, Fig. 3b) and the soluble nuclear compartment (S3, Fig. 3c), and the concomitant increase of TRβ1 levels in the insoluble nuclear compartment (P3, Fig. 3d). Since, the experiments presented in Figure 3 include both TRβ1 movement from one cellular fraction to another and TRβ1 degradation, whereas experiments presented in Figure 1 include only TRβ1 degradation, these two figures cannot be directly compared.
It is tempting to speculate that soon after T3 treatment, the increased TRβ1 population in the insoluble nuclear fraction could be implicated in transcriptional regulation of target genes. This assumption is coherent with many rapid T3-induced events: nuclear transport of TRβ1 (18), recruitment of TRβ1 to the promoters of target genes (30), and induction of target genes (31). Further, TRβ associates with the nuclear matrix in a ligand-dependent fashion (17), the nuclear matrix is linked to gene transcription (32 –36), and the proteasome co-exists with NRs and coactivators in nuclear matrix-associated foci after ligand stimulation (37). Therefore, TRβ1 increasing in the insoluble nuclear fraction could be recruited to the nuclear matrix to regulate target gene transcription, and after that, this population would be degraded. To verify these hypotheses and expand our comprehension of the molecular mechanisms of TH action, TRβ1 population associated with the nuclear matrix should be separated from TRβ1 population associated with the chromatin. Further, because phosphorylation of TRs might orchestrate changes in TR localization (38) and that phosphorylation seems to stabilize TRβ1 (26,27) and to increase its transcriptional activity (27,39 –41), it could be involved in the rapid increase of TRβ1 in the insoluble nuclear fraction and could protect transiently this population of receptors from degradation. Thus, post-translational modification of TRs in each fraction should also be explored in future studies.
Recognition of target proteins by the ubiquitin-proteasome pathway is mediated through specific motifs that are not clearly understood. Many rapidly degraded regulatory proteins contain PEST motifs (3,24). We demonstrated that a mutation in the putative PEST motif of TRβ1 does not modify its proteasomal degradation (Fig. 4). Other NRs [RORα (42), ERα (10), and RXR (43)] also contain PEST motifs that are not involved in their degradation. Thus, alternate motifs should be searched for.
Conclusion
This study leads to a better comprehension of the kinetics of TRβ1 degradation by showing that TRβ1 degradation is atypical due to the co-existence of different TRβ1 populations that are targeted for degradation by the proteasome pathway with different efficiencies. Moreover, our results suggest a movement of TRβ1 from the cytoplasm toward the nucleus and/or a subnuclear movement of TRβ1 between the soluble nuclear fraction and the insoluble nuclear fraction, in the presence of T3, and the existence of TRβ1 associated to chromatin and/or nuclear matrix. The finding that stable and unstable forms of TRβ1 co-exist suggests that the rate of degradation of TRβ1 protein might be related to specific functions of these TRβ1 populations. Thus, in future studies it will be necessary to better characterize each TRβ1 population and to investigate the link between TR functions and proteasome-mediated proteolysis. This is relevant to better understand TH action.
Footnotes
Acknowledgments
This work has been supported, in part, by grants from the Canadian Institutes of Health Research (CIHR), the Faculté de médecine et des sciences de la santé de l'Université de Sherbrooke, and the Centre de recherche clinique Étienne-Le Bel, an FRSQ research center. M.-F.L. is the recipient of a senior clinician-investigator scholarship from the Fonds de la recherche en santé du Québec (FRSQ), T.F. is the recipient of a Post-doctoral Fellowship from Pfizer Canada Inc. and the Département de médecine de l'Université de Sherbrooke, and M.B. of master's degree scholarships from FRSQ and CIHR.
Disclosure Statement
The authors declare that they have no competing financial interests.