
Abstract
Background:
Increasing evidence suggests that thyroid hormones, L-thyroxine (T4) and 3,3′,5-triiodo-L-thyronine (T3), are modulators of the immune response. In monocytes, macrophages, leukocytes, natural killer cells, and lymphocytes, a wide range of immune functions such as chemotaxis, phagocytosis, generation of reactive oxygen species (ROS), and cytokine synthesis and release are altered under hypo- and hyperthyroid conditions.
Summary:
Hyperthyroidism decreases the proinflammatory activities of monocytes and macrophages, whereas enhancement of phagocytosis and increased levels of ROS may occur during hypothyroidism. The expression of proinflammatory molecules such as macrophage inflammatory protein-1α and interleukin-1β increases in hypothyroidism. However, in Kupffer cells, proinflammatory activities such as the respiratory burst, nitric oxide synthase activity, and tumor necrosis factor-α expression may result from increased T3 levels. Thyroid hormones also affect natural killer cell activity and cell-mediated immune responses. Still, for many immune cells no clear correlation has been found so far between abnormally high or low T3 or T4 levels and the effects observed on the immune responses.
Conclusions:
In this review we outline the contributions of thyroid hormones to different aspects of innate and adaptive immune responses. The relationship between thyroid hormones and immune cells is complex and T3 and T4 may modulate immune responses through both genomic and nongenomic mechanisms. Future studies of the molecular signaling mechanisms involved in this cross-talk between thyroid hormones and the immune system may support development of new strategies to improve clinical immune responses.
Introduction
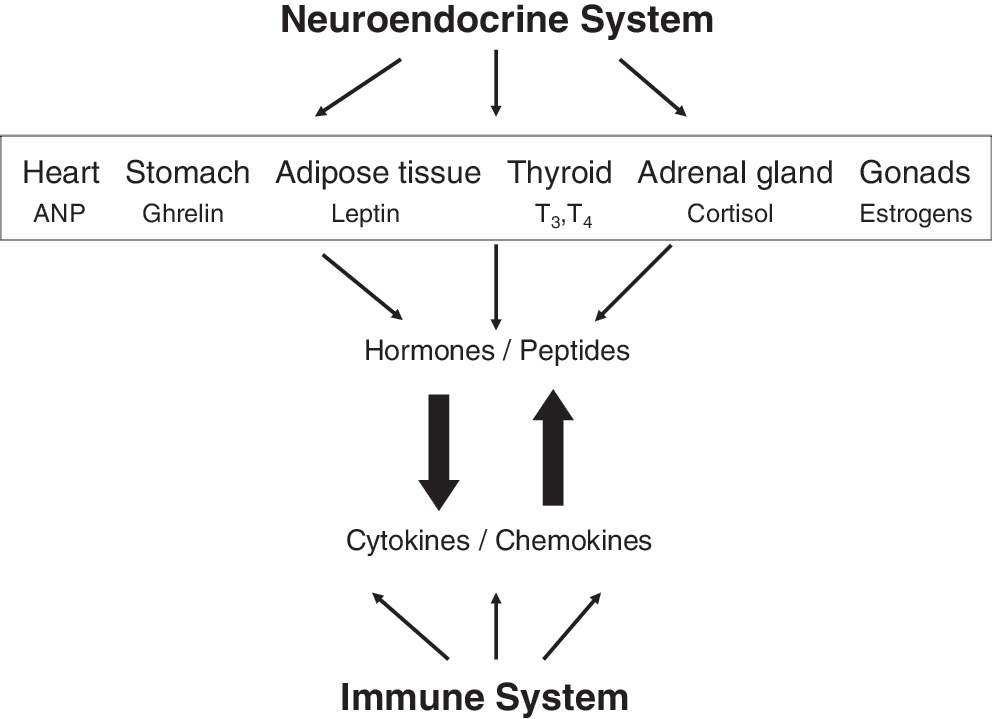
Neuroendocrine and immune system cross-talk. Depicted are certain endocrine organs and nonendocrine organs, such as the stomach and adipose tissue, with their hormone or peptide products that are known to interact with the immune system. In turn, the immune system may modulate the function of the same tissues and organs and their products through the release of cytokines and chemokines.
3,3′,5-Triiodo-L-thyronine (T3) and L-thyroxine (T4) modulate specific immune responses, including cell-mediated immunity, natural killer cell activity, and the antiviral action of IFN and proliferation of T and B lymphocytes (15). In addition to the production of cytokines, chemokines, and eicosanoids, such as prostaglandins and leukotrienes, the cells of the immune system produce several hormones or hormone-like molecules (16), and the presence of thyroid hormone (T3) has been reported in immune cells such as monocytes, granulocytes, natural killer cells, mast cells, and lymphocytes (17 –20). The level of thyroid hormone in these cells was decreased by treatment with thiamazole, an antithyroid drug (18), or increased in cells obtained from animals treated with thyrotropic hormone (TSH) (20).
However, our understanding of the connections between thyroid hormones and the immune system is still rather incomplete, because each of these subjects intrinsically is very complex and their interaction is even more complicated. The body of information on the relationship between thyroid hormones and immunity is relatively large, and the data available are sometimes difficult to interpret or even contradictory. This article reviews current evidence supporting the contribution of thyroid hormones to the modulation of immune activities such as chemotaxis, phagocytosis, respiratory burst, cytolytic activity, and cytokine synthesis in monocytes, macrophages, leukocytes, natural killer cells, and lymphocytes.
Thyroid Hormone Actions
Thyroid hormones have diverse effects on striated muscle, heart and blood vessels, bone, liver, and the central nervous system. These are mediated by specific thyroid hormone receptors (TRα and TRβ) that are nuclear in location or may translocate from the cytoplasm into the cell nucleus to regulate gene expression (21 –24). Although both T3 and T4 enter cells, only T3 is the ligand for TRs. The T3-TR complex binds to nuclear thyroid hormone response elements—specific DNA sequences found in the regulatory regions of target genes—causing conformational changes of the chromatin that appear to render the genes accessible to the transcriptional machinery (25). Resulting gene products in turn mediate classic genomic thyroid hormone actions on many homeostatic functions of cells and on morphogenesis during embryonic development. Genomic actions therefore may require hours to manifest. In contrast, a number of thyroid hormone effects occur rapidly (seconds to minutes) and are unaffected by inhibitors of transcription or translation, the so-called nongenomic actions of thyroid hormones (21,26 –32). Most nongenomic thyroid hormone effects are initiated at the plasma membrane, but in some cases the effects may have their origin in the cytoplasm or in organelles. These effects include modulation of ion and glucose transport through activation of protein kinases C (PKC) and A, mitogen-activated protein kinases (MAPKs) and in particular the extracellular-regulated kinases 1/2 (ERK1/2) and phospholipases C and D (30 –32). Some of the authors of the present article have shown that a structural protein of the plasma membrane, integrin αvβ3, bears a cell surface receptor for iodothyronines (33 –35). Via this receptor the hormone can activate both the ERK1/2 and phosphatidylinositol 3-kinase (PI3K) pathways. Downstream consequences of ERK1/2 activation include intracellular protein trafficking, angiogenesis, and tumor cell proliferation (35 –39). The ability of thyroid hormone analogs to interact with αvβ3 provides another perspective on the mechanisms by which such compounds modulate cellular defenses.
Altered Thyroid Status and Immune Responses
Hyperthyroidism is a pathological condition due to excessive production and release of thyroid hormones from the thyroid gland. It can be caused by thyroid adenoma or multinodular goiter, and commonly by the antibody-mediated Graves' disease; hyperthyroidism is only rarely the result of abnormalities of the hypothalamic-pituitary pathway (40,41). Dysregulated T lymphocytes are involved in Graves' disease; these are mainly CD4+ Th2 cells, although Th1 cells can also contribute (41).
Hypothyroidism is the result of insufficient production of thyroid hormones by the thyroid gland (42) and is classified according to the point of origin of the condition in the hypothalamic-pituitary-thyroid axis. Primary hypothyroidism reflects thyroid gland failure, most commonly due to Hashimoto's thyroiditis, in which the immune system attacks the thyroid gland. Hashimoto's disease is a common T cell-mediated autoimmune disease, characterized by infiltration of the thyroid gland with mononuclear cells, B-cells, and T lymphocytes, mainly of the CD4+ Th1 subtype. A less common type of hypothyroidism (secondary hypothyroidism) occurs when the pituitary gland does not release TSH. Finally, in tertiary hypothyroidism the origin of the disease is a disordered relationship between the hypothalamus and pituitary gland (43).
Hypothyroidism can also result unintentionally from the use of thioamide drugs, such as propylthiouracil (PTU) (44) or methimazole, for treatment of thyrotoxicosis. In addition, PTU has been associated with suppressive or stimulatory events in the immune response (45); depending on the duration of treatment, the same dose of PTU can either stimulate or suppress antibody production in rats (46). Dose-dependent hypo- or hyperfunction of suppressor T cells have both been observed with PTU (47). Methimazole also affects host defenses and can repress the activity of peritoneal as well as alveolar macrophages (48). We would note parenthetically here that PTU is the subject of a U.S. Food and Drug Administration physician advisory because it can induce hepatocellular failure and is no longer a drug of choice for clinical management of most hyperthyroid states.
Induction of hypothyroidism by different experimental means leads to spleen and lymph node involution as well as to a decrease in humoral and cell-mediated immune responses (49,50). Severe clinical hypothyroidism due to chronic autoimmune thyroiditis is accompanied by a drastic decrease in lymphocyte function; gradual improvement in lymphocyte function has been documented after euthyroid status has been re-established (51). In agreement with these observations is the recovery after T4 treatment of the T-cell lymphoproliferative response in chronically stress-exposed mice that become hypothyroid (52). Low concentrations of T3 (10−9 M) and T4 (10−7 M) can stimulate T-cell proliferation (53); these total hormone concentrations correspond to the physiological levels of each hormone. Table 1 summarizes the major effects of hyperthyroidism and hypothyroidism on several aspects of immune function. The table reveals that although hypo- and hyperthyroidism produce opposite effects on some parameters (proinflammatory markers, lymphocyte proliferation, and antioxidant capacity), other immune functions do not show any simple monotonic response to changes in thyroid hormone concentrations. The effect clearly depends very much on the precise cellular context and clinical conditions.
↑, increase; ↓, decrease; –, no change.
Activation of T lymphocyte subclasses, reduction of natural killer cells and decrease in T CD4 lymphocyte responsiveness occur in severe hypothyroidism (54), and in clinical cases of hypothyroidism the spontaneous migration of polymorphonuclear leukocytes (PMNs) was found to be impaired, when compared to healthy subjects (55). In contrast, a study of PMNs from hyperthyroid patients revealed normal migration and chemotactic activity (56). Studies conducted on hyperthyroid and hypothyroid patients indicate that endogenous and exogenous thyroid hormones increase oxidative metabolism (57 –61). PMNs from thyroidectomized patients were found to have lower levels of resting O2 − generation, both in the initial hypothyroid condition and after restoration of euthyroidism by T4. PMNs harvested during the hypothyroid state also showed an impaired ability to generate O2 − after stimulation with N-formyl-Met-Leu-Phe, but not with phorbol 12-myristate 13-acetate (58,59,61).
Contradictory results are reported in the literature with regard to the effect of hyperthyroidism on immunity, since both enhancing and suppressing effects on humoral and cellular immune responses have been described (62 –65). Induction of hyperthyroidism in rats did not affect the ratio of helper to suppressor T lymphocytes (47). During treatment of hyperthyroid Wistar rats with T3 (25 μg/day for 7–12 days), it was found that antibody production was significantly reduced when compared to normal rats immunized with the same antigen (sheep red blood cells), suggesting that hyperthyroidism in these experimental settings decreases the antibody response (66). These authors found the opposite effect, an increase in antibody response in Wistar rats, after PTU treatment for the same period (46). In rats immunized with sheep red blood cells and subsequently treated for 4 weeks with T4 (25 μg/kg per day), the primary antibody response to sheep red blood cells was higher than in control animals (67). These results indicate that the antibody production is affected by thyroid hormone levels.
Studies carried out on development and function of lymphocytes in mice with genetic defects involving the production of thyroid hormones or their nuclear receptors indicate that thyroid hormone may not be essential for development of a normal lymphocyte response. It has been proposed that thyroid hormones are involved in maintaining immune system homeostasis in response to environmental changes or stress-mediated immunosuppression (68). However, no alteration of humoral or cell-mediated immune response has been reported in dwarf mice with defects in genes encoding thyroid hormones or other anabolic hormones, or in thyroid hormone-deficient hyt/hyt knockout mice (64).
Macrophages
The principal mechanisms by which monocytes and macrophages elicit innate immunity include chemotaxis, phagocytosis, and the generation of reactive oxygen species (ROS) such as hydrogen peroxide and superoxide (69,70). Thyroid hormone-induced effects in macrophages are correlated with the hypo- or hyperthyroid states. In hyperthyroid rats (administration of 500 mg T4 and 125 mg T3 kg−1 body weight for 7 days), immune system stimulation with thioglycolate and/or Bacillus Calmette-Guerin revealed that monocyte chemotaxis, phagocytic activity, and hydrogen peroxide release by macrophages were suppressed. In hypothyroid rats, conversely, macrophage phagocytosis and ROS release were both enhanced, whereas monocyte migration was not affected (71). Macrophages are the main source of several proinflammatory cytokines, including IL-1, IL-6, and TNF-α (72). During streptococcal cell wall-induced inflammatory response in a strain of rats frequently used as a model for rheumatoid arthritis, the magnitude of macrophage inflammatory response depends on the hypo- or hyperthyroid condition (73). In particular, in rats rendered hypothyroid by PTU, streptococcal cell wall administration stimulates expression by macrophages of pro-inflammatory markers such as the chemokine macrophage inflammatory protein-1α (MIP-1α) and IL-1β. In contrast, in rats with T4-induced hyperthyroidism the expression of the same markers was inhibited. In this connection, the increased expression in hypothyroidism of several proinflammatory markers seems to be positively coupled to an increase in mRNA of MIP-1α and IL-1β as measured by northern blot analysis (73). These findings infer that high circulating levels of thyroid hormone may oppose several proinflammatory mechanisms in which monocytes and macrophages are involved.
Chemotaxis represents an important function by which macrophages confront certain pathogens (74). Strenuous exercise stimulates murine macrophage chemotaxis and the exposure in vitro of these cells to T3 and T4 concentrations similar to those found in plasma after exercise (T3, 2.3 nM; T4, 84 nM) reproduce this effect (75). These data support a role for thyroid hormones in exercise-induced chemotaxis.
The decrease of thyroid hormone production and release in aged human subjects (76) contributes to the immune dysfunction associated with normal aging. Recently, El-Shaikh et al. (77) have shown that the infusion of T4 (0.2–5 μg) in BALB/c mice for 30 days induces an increase in the total number of peripheral blood leukocytes, as well as increased cellularity of thymus, spleen, and lymph nodes and increased macrophage phagocytosis. The discovery that T4 elicits such responses is in agreement with the observation that in old animals the administration of T4 promotes regrowth of the thymus, recovery of its endocrine function, and the repair of age-related immune dysfunction (78). Macrophages are generally derived from peripheral blood monocyte migration and differentiation; however, in organs such as liver and bone there are resident macrophages known as Kupffer cells and osteoclasts, respectively (79,80). Kupffer cells are a heterogeneous group of cells derived from local precursors that are morphologically distinguishable from mature lineage elements (81). The administration of T3 to rats (0.1 mg/kg for 3 consecutive days) has been found to increase several proinflammatory functions mediated by Kupffer cells, including the respiratory burst, nitric oxide synthase (iNOS) activity, and TNF-α expression. T3-induced TNF-α expression, in turn, involves IκB-α phosphorylation and activation of nuclear factor κB (NFκB). One study of such functions (82) relied upon very high doses of T3 that were 70 times greater than physiological values. However, other authors have established the ability of T3 to increase marker events in Kupffer cells, such as the DNA binding activity to NFκB, the expression of TNF-α and IL-10, and oxidative stress (83,84). Increased plasma levels of T3 are associated with bone mass loss, but the mechanism by which thyroid hormone affects bone turnover remains unclear. It has been suggested that T3 indirectly stimulates osteoclastic bone resorption through the release of soluble mediators from osteoblasts (85). In fact, T3 (10−12–10−8 M) directly stimulates the production of IL-6, and also IL-1-induced IL-6 production in osteoblast-lineage cells. T3 (1–100 nM) induces cytokine release in osteoclast hOb cell lines, such as MG63 and SaOs-2, as well as in human bone marrow stromal cells, and markedly increases the expression and release of IL-6 and IL-8 in both osteoclast and bone marrow stromal cells (86). These findings implicate IL-6 as a mediator of the contribution of thyroid hormone to bone resorption (87). Hoffman et al. (88) showed that 3 weeks of daily administration of T4 (250 μg/kg) in rats led to a significantly lower bone mineral density compared to untreated controls; interestingly, the T4-induced reduction of bone mass was blocked by a proprietary pharmacological inhibitor of integrin αvβ3 and this is in line with the finding that integrin αvβ3 contains a plasma membrane receptor of thyroid hormones (33 –37) (Fig. 2).
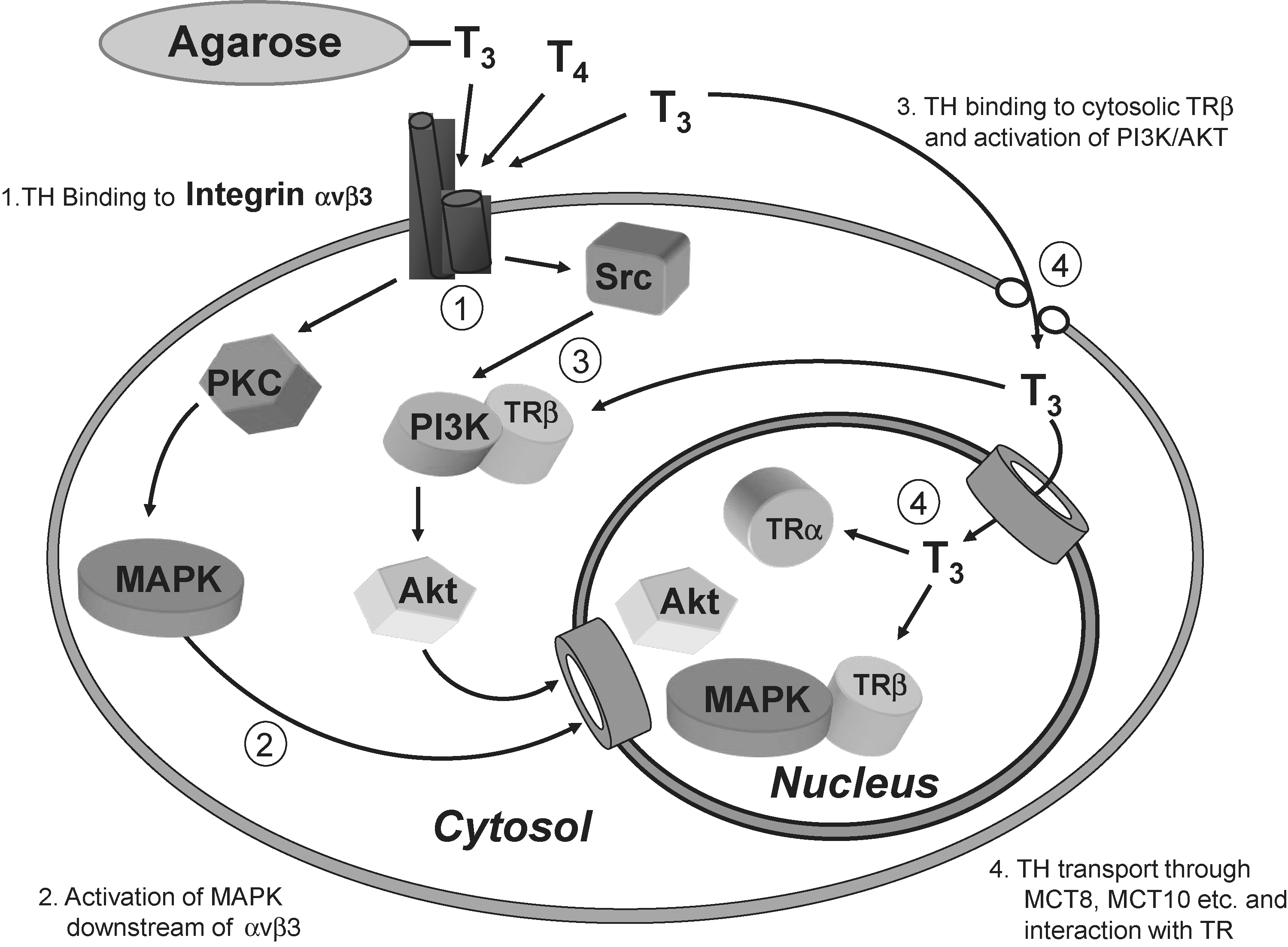
Thyroid hormone receptors in a standard cell type and the different signal transduction pathways activated. The αvβ3 integrin receptor is located at the plasma membrane, whereas the classic nuclear receptors TRα1 and TRβ1 may shuttle between the nucleus and the cytosol. The use of membrane-impermeant hormone conjugates such as agarose-3,3′,5-triiodo-L-thyronine (T3) permits distinction between cytoplasmic (cytosolic) actions and effects initiated at the plasma membrane.
Physical exercise-related stress gives rise to high plasma concentrations of so-called stress hormones, including T3 and T4 (89,90). Moderate exercise stimulates the immune system, whereas strenuous exercise impairs immune response (91 –94). Exposure of macrophages in vitro to T3 concentrations similar to those found in the plasma after exercise (1.5 ng/mL) did not affect the phagocytic activity of these cells, whereas phagocytosis was stimulated by physiological concentrations of T4 (95). However, previous studies on the effects of thyroid hormones on macrophage phagocytosis have shown that T3, under different experimental conditions, can promote the phagocytic activity of these cells (96). Since phagocytosis can be studied using either opsonized cells or nonopsonized inert particles, it is possible that the different responses elicited by thyroid hormones may depend on the type of antigen phagocytosed.
There is evidence that infiltration of airways by inflammatory cells plays an important role in the development of bronchial asthma (97,98). In this process cells such as monocytes and macrophages contribute to the release of several inflammatory mediators such as prostaglandins, leukotrienes, and superoxide anions (99). Nishizawa et al. (100) have shown that in human macrophages isolated from bronchoalveolar lavage fluid, the exposure of cells to T4 (10–50 ng/mL) stimulates the release of superoxide anion; this observation is in good agreement with studies showing that the hyperthyroid state increases airway reactivity and can exacerbate bronchial asthma (101). The dangerous consequences of ROS generation can be limited by several antioxidant mechanisms, including the expression of the superoxide dismutase (SOD) enzyme (102). Interestingly, in aged rats the induction of a hyperthyroid condition by thyroid hormone treatment leads to a decrease in Cu/Zn SOD-1 activity, whereas progressive hypothyroidism leads to an increase in SOD activity (103,104). In the monocytic cell line U937, nanomolar concentrations of T3 inhibit the activation of the SOD-1 promoter induced by phorbol 12-myristate 13-acetate or by free radical generators such as paraquat (105). The ability of thyroid hormones to induce a suppressive effect on the regulation of the SOD-1 promoter may in part explain the relationship between the excess of oxidative stress and the hyperthyroid condition.
Leukocytes
Thyroid hormones can promote oxidative stress by stimulating several enzyme systems, including cytochrome P450, myeloperoxidase, and plasma membrane NADPH oxidase, which are responsible for superoxide anion generation and bacterial killing (106). ROS generation is enhanced in the hyperthyroid state (107,108) and mitochondrial oxygen consumption of PMNs of patients affected by Graves' disease is higher than in normal individuals (59). In euthyroid subjects, the oral administration of T3 (60 μg/day) for 7 days induces an enhancement of ROS generation by mononuclear cells and by PMNs (109). This effect has been positively correlated with an increase of intracellular levels of meta-tyrosine and ortho-tyrosine, two well-known markers of oxidative damage of proteins and amino acids (110). The ability of T3 to stimulate the respiratory burst of PMNs through ROS generating systems, such as myeloperoxidase and NADPH oxidase, has been reported (111), and additional experimental evidence is available to support the relationship between thyroid hormone and ROS production (112 –114). This is also confirmed by the enhancement of superoxide generation induced by N-formyl-Met-Leu-Phe in PMN cells in hyperthyroid patients (59), which may be responsible for the exacerbation of bronchial asthma observed in these patients (101,115).
Interestingly, Mezosi et al. (116) have demonstrated that T4 and T3 act on human PMNs to generate superoxide anions and to increase myeloperoxidase activity in a rapid, nongenomic manner. The thyroid hormone-induced ROS generation can be totally prevented by inhibitors of PKC (such as calphostin C and Ro-32-0432) or by the calcium chelator BAPTA. Partial inhibition of the thyroid hormone effect was obtained with pertussis toxin (116). These data suggest that hormone-induced ROS generation in human PMNs could be mediated by a membrane-bound receptor for thyroid hormones.
The finding that the integrin αvβ3 acts as a plasma membrane receptor for thyroid hormones (33) provides a new perspective on the role of thyroid hormones in the modulation of cellular defense. Lin et al. (117) have shown that T4 potentiates the antiviral effect of IFN-γ by a nongenomic mechanism; this finding was among the first to support the idea that thyroid hormone may play an important role in cellular defense mechanisms via nongenomic pathways. In HeLa cells, T4 at 10−7 M, corresponding to the physiological total hormone concentration, stimulates IFN-γ-induced human leukocyte antigen DR expression, a marker of immune status (118). This action is STAT1α-dependent. Interestingly, the treatment of cells with T4-agarose, a membrane-impermeant analog of T4, also caused activation of STAT1α, demonstrating more than a decade before identification of the integrin receptor that the T4 effects are mediated by a cell membrane hormone binding site. Thyroid hormones (10−7 M T4, or 10−9 M T3) potentiate the antiviral effect of IFN-γ in HeLa cells (119); the effect is significant within 2 hours, provided that the cells have been previously exposed to IFN-γ for 20 hours. In this system T4 is 10-fold more effective than T3. A 120-fold increase in the response to IFN-γ was observed in human (BG-9) fibroblast cells when they were pretreated for 3 hours with T4, 0.5 × 10−10 M (117). Lin et al. (120) have shown that in HeLa cells T4 promotes Ser-727 phosphorylation of STAT1α by an ERK1/2-dependent mechanism, a specific step that potentiates transduction of upstream signals of IFN-γ by this phosphorylated STAT protein. These MAPK (ERK1/2)-mediated actions of thyroid hormone may also be subject to inhibition by pertussis toxin (120), as were the thyroid hormone effects on PMN ROS generation (116), to which reference was made earlier. These short-term effects induced by thyroid hormone are all consistent with the involvement of nongenomic mechanisms.
Natural Killer Cells
Natural killer cells, through secretory and nonsecretory mechanisms, are essential in the initiation of the adaptive immune response during viral infections or tumor growth (121 –123). Increasing evidence suggests that thyroid hormones play a key role in the modulation of natural killer cell activity and provides new insights into the mechanisms by which the endocrine system affects the biological activity of the immune system (124,125). Both innate and adaptive immunity are often dysregulated in older people and such changes can increase susceptibility to infections, cancer, and autoimmune diseases (126). Dysregulation may to some degree reflect an age-related decline in thyroid hormone levels; in fact, the cytolytic activity of natural killer cells isolated from older human subjects can be restored by T3 (10−7 M) (127). It has also been shown in animal models that a low-protein diet depressed natural killer cell activity; this was not observed when the protein-deficient diet was supplemented with T3 (128). Conversely, the in vivo administration of T3 alone had no effects on the cytolytic activity of avian natural killer cells (129). Thyroxine also plays a role in the modulation of natural killer cell activity; the administration of T4 markedly increased the cytolytic activity of murine natural killer cells (130,131). It remains to be established whether these effects involve specific T4-nongenomic signaling or simply are due to local deiodinase activity that converts T4 to T3.
It is well known that cytokines such as IL-2, IL-12, IL-15, IL-18, and IFN-γ stimulate human natural killer cell proliferation and activation (132). T3 has been shown to enhance the responsiveness of avian natural killer cells to IL-2, as well as the expression of the IL-2 receptor (133,134). This action of the hormone is biphasic, in that addition of low levels (0.1 ppm) of T3 to the diet results in enhancement of IL-2 receptor expression, whereas a further increase in T3 concentration in the diet to 1 ppm decreases such expression. The ability of T3 (1–100 nM for 3 days) to induce expression of the IL-2 receptor has also been reported in other immune cells, such as the peripheral blood mononuclear cells and YT cells (an IL-2-independent natural killer-like cell line) (68). The activity of natural killer cells is usually stimulated by IFN-γ, and administration of low doses of T4 (0.01–0.1 μg/mL) increased the stimulatory effects of IFN-γ on murine natural killer cells (131). A similar increase in avian natural killer cytolytic activity was obtained by stimulation with thymulin, a hormone able to promote the expression of IFN-γ receptors (135). However, it should be remembered that the findings reported in references 133–135 were obtained in avian cells and these findings may not be transferable directly to mammalian cells.
Lymphocytes
Studies on the roles of thyroid hormones in adaptive immunity have shown that human hypothyroidism, as well as pharmacologically induced or surgical hypothyroidism in rodents, is associated with a decrease in thymic activity. This deficiency can usually be remedied by the administration of thyroid hormones (50,63). A positive correlation between hypothyroid status and spleen and lymph node involution, as well as a decrease in humoral and cell-mediated immune responses, has been reported (136,137). Decreased responsiveness of peripheral blood lymphocytes from hypothyroid animals to standard mitogens, such as phytohemagglutinin and concanavalin A, has been recognized for many years (138,139). Although much evidence suggests that thyroid hormone modifies lymphocyte activities, the magnitude of the hormone effects seems to depend on the source of lymphoid cells, and the physiological mechanisms involved have not been elucidated to-date (140,141). Lymphocyte homing is a multistep process via a specialized pathway that is triggered by interaction of T cells with selectins, chemokines, integrins, and other extracellular matrix molecules (142). In Balb/c mice the administration of T3 modulates the homing of recent thymic emigrants (RTEs) to peripheral lymphoid organs, giving rise to an increase in the abundance of CD4+ RTEs and CD8+ RTEs in subcutaneous and mesenteric lymph nodes (143), whereas CD4+ RTE cells are relatively decreased in the spleen. Moreover, the expression of extracellular matrix proteins such as laminin and fibronectin—proteins that play a role in T-cell migration—increases in lymph nodes, but not in the spleen, after intrathymic administration of T3 (143). In T cell hybridomas, induction of the T-cell receptor (TCR)/CD3 complex leads to apoptosis (144); in such cells glucocorticoids inhibit TCR/CD3-mediated apoptosis. However, in cells in which TCR/CD3 is not induced, glucocorticoids alone are able to induce apoptosis, whereas thyroid hormone does not affect apoptosis (144).
Little is known about effects of thyroid hormone on the humoral immune response. In some studies, this response is enhanced after thyroid hormone treatment (129,145,146), and thyroidectomy suppresses this immune response (145). However, contradictory results have also been reported, describing a suppressed antibody response after thyroid hormone treatment (147,148) and an enhanced humoral immune response after antithyroid drug exposure (46). The authors of one study reported that thyroid hormone does not have any effect on the humoral response in domestic fowl (149). There is evidence that T4 promotes immunoglobulin M (IgM) production in mouse lymphocytes and, as well, stimulates the humoral response to immunization with Salmonella typhi (146). The mechanism involved in T4-induced IgM production is still unknown.
Reports concerning the intracellular signaling pathway(s) involved in the modulation of T cell proliferation by thyroid hormone describe different signaling mechanisms in normal and tumoral T lymphocytes. Proliferation of normal lymphocytes induced by T3 (1 nM) is mediated by conventional PKCs and calcium-dependent mechanisms, whereas in BW5147T lymphoma cells the hormone-induced effect is obtained through activation of nonconventional Ca2+-independent PKCs, such as the isoenzyme PKCζ or by iNOS (41); similar results were obtained using T4 at 100 nM. The ability of thyroid hormones at high concentrations to stimulate murine leukemia cell proliferation has also been reported (147). Recently, it was shown that the effects of T3 and T4 on T cell proliferation depends on both genomic and nongenomic mechanisms (150); signal transduction pathways initiated at the plasma membrane activated PKCζ and caused phosphorylation of ERK1/2, and subsequently activation of NFκB. In contrast, iNOS induction depended on the nuclear receptors (150).
However, physiological concentrations of thyroid hormone induce apoptosis in Jurkat human T lymphoma cells (151). Treatment of T lymphocytes with T3 or T4 gave rise to enhancement of apoptosis and reduction of mitochondrial transmembrane potential (Δψ), as well as to decreased expression of antiapoptotic protein Bcl-2 and an increase in ROS generation; all of these markers are usually associated with apoptotic cell death (151). Interestingly, T lymphocytes derived from patients with Graves' disease exhibit increased apoptosis compared to cells obtained from euthyroid individuals (152), suggesting that thyroid hormone can induce apoptosis in human lymphocytes in vivo, as well as in vitro.
The expression of myeloid cell leukaemia-1 protein, an antiapoptotic member of the B-cell lymphoma-2 family, increases in TR-expressing HEK293 cells after 6 hours of incubation with T3 (10−7 M). This occurs by a nongenomic mechanism that involves both PI3K and TRβ1 (153). T3 can decrease the amount of DNA laddering as well as the number of TUNEL-positive cells in infarct-border areas after myocardial infarction in rats, suggesting that thyroid hormone has a protective function during ischemia-induced apoptosis (154). We conclude that thyroid hormones can be either antiapoptotic or proapoptotic, depending on the cell type under investigation.
Dendritic cells are highly specialized antigen-presenting cells that recognize, process, and present antigens to T cells for the induction of antigen-specific immune response (155). In immature as well as lipopolysaccharide-induced and bone marrow-derived dendritic cells—in which the presence of TRα and TRβ in the cytoplasm has been demonstrated—exposure to physiological concentrations of T3 induces expression of specific dendritic cell maturation markers. These include major histocompatibility complex II, CD80, CD86, and CD40 (156); IL-12 secretion is also enhanced. Certain thyroid diseases are examples of endocrine autoimmune dysfunction, and it is possible that the local production of T3 within the thyroid gland amplifies dendritic cell function/maturation and thus contributes to immune-mediated pathological conditions of the gland (157).
Concluding Remarks
Thyroid hormones influence specific immune responsiveness, as well as several aspects of innate and adaptive immunity. The relationship between thyroid hormone and immune cells is complex and additional insights are needed into the molecular signaling mechanisms involved in the cross-talk between thyroid hormone and immune function. Still, from the limited knowledge currently available it is possible to draw some conclusions: (i) T3 and T4 modulate immune responses through both genomic and nongenomic mechanisms and, at physiological concentrations, support the basal function of immune cells. A schematic representation of genomic and nongenomic signaling by thyroid hormones is shown in Figure 2. (ii) The functions of immune cells, including generation of ROS, chemotaxis, phagocytosis, and cytokine synthesis and release, are affected by thyroid gland dysfunction, in the form of either hypo- or hyperthyroidism. (iii) Innate and adaptive immune responses undergo significant changes during aging and in hypothyroidism, although the exact nature of the changes in hypothyroidism requires further clarification. (iv) Physiological concentrations of T3 stimulate the production and release of cytokines, as shown by the work of Shih et al., who found that 28 genes related to inflammation are upregulated by T3 in TRα-1-overexpressing cells (158). (v) The capability of thyroid hormones to potentiate the antiviral action of IFN-γ and a number of subsequent studies of thyroid hormone actions on immune cells reviewed above suggest that clinical manipulation of thyroid hormone levels, at least in the short-term, could be applied therapeutically to modulate immune functions.
Footnotes
Acknowledgments
Dr. Terry Davies kindly provided very helpful comments and criticisms during the preparation of the article. Financial support from the Italian Ministry for Education, University and Research, General Management for International Research, is gratefully acknowledged.
Disclosure Statement
The authors declare that no competing financial interests exist.