
Abstract
Background:
The thyroid stimulating hormone receptor (TSHR) is the key regulator of thyrocyte function. The gene for the TSHR on chromosome 14q31 has been implicated as coding for the major autoantigen in the autoimmune hyperthyroidism of Graves' disease (GD) to which T cells and autoantibodies are directed.
Summary:
The TSHR is a seven-transmembrane domain receptor that undergoes complex posttranslational processing. In this brief review, we look at the genetics of this important autoantigen and its influence on a variety of tissue functions in addition to its role in the induction of GD.
Conclusions:
There is convincing evidence that the TSH receptor gene confers increased susceptibility for GD, but not Hashimoto's thyroiditis. GD is associated with polymorphisms in the intron 1 gene region. How such noncoding nucleotide changes influence disease susceptibility remains uncertain, but is likely to involve TSHR splicing variants and/or microRNAs arising from this gene region. Whether such influences are confined to the thyroid gland or whether they influence cell function in the many extrathyroidal sites of TSHR expression remains unknown.
Introduction
The TSH receptor

Model of the TSHR. The TSHR first appears on the plasma membrane as an intact holoreceptor. This model shows the seven transmembrane domains and the large extracellular domain. The extracellular domain consists of the 10 leucine-rich repeats in the ectodomain, which is the major site of TSH binding, and the 50 amino acid cleaved region. Reproduced with permission from Rees Smith et al.
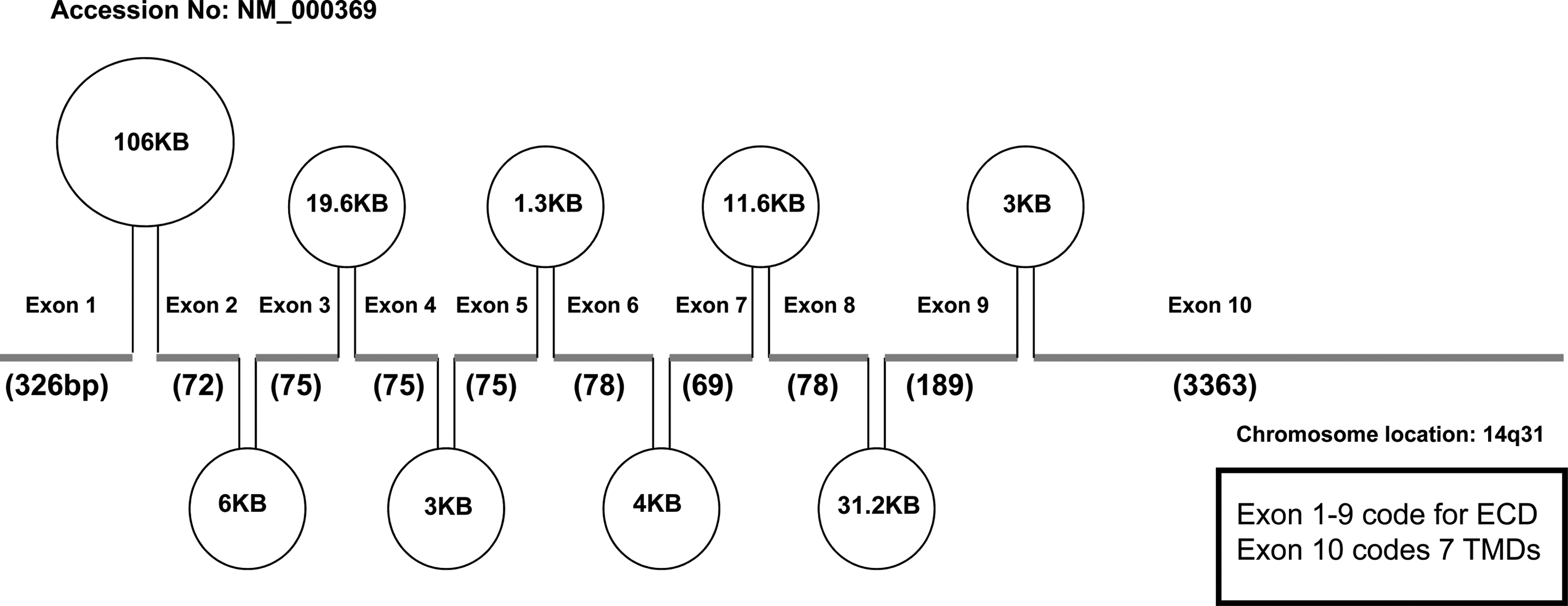
The exon–intron arrangement of the human TSHR gene. This diagram indicates the exon and intron arrangement of the human TSH receptor gene on chromosome 14q31. The exons with their sizes are indicated with the thick line segments, and introns within the exons are indicated with their sizes as ovals. The first 9 exons of the TSHR gene code for the large extracellular domain and exon 10 codes for the seven transmembrane regions.

Posttranslational processing of TSHR. The holoreceptor undergoes cleavage and loses residues ∼316–366. This results in the formation of a two subunit structure (α & β) connected by disulfide bonds and is referred to as the cleaved TSHR. Upon reduction the α is thought to shed from the cell surface leaving the membrane-anchored β subunits in excess.
TSH, thyroid stimulating hormone.
Graves' disease
GD is an autoimmune thyroid disease (AITD), which together with HT, are immune disorders characterized by the infiltration of thyroid tissue by immune effector cells including the presence of thyroid-antigen-specific T cells and the production of autoantibodies to well-defined thyroidal antigens such as thyroid peroxidase (TPO), thyroglobulin (Tg), and the TSHR (17). Both diseases are seen more often in women than in men with a female:male ratio of at least 5:1 and have a population prevalence of ∼10% (18). A genetic determinant to the susceptibility to AITD was first suspected because of familial clustering of the diseases, a high sibling recurrence risk of >16 (19), the familial occurrence of thyroid antibodies (TAbs) (20,21), and the 30% concordance in disease status between identical twins (22). Since both GD and HT may occur within the same family, this also suggested the existence of susceptibility genes common to both diseases.
The dawn of AITD genetics
The production of TAbs to TPO and Tg often precedes the development of clinical AITD, and TAbs have been widely used to show the population at most risk for the development of AITD. For example, in women who were positive for TAbs, and who had an abnormal TSH, the annual risk of developing hypothyroidism was 2%–4% in a number of different studies (18). As mentioned earlier, genetic susceptibility to the production of TAbs was first suggested by Hall and Stanbury (20). Their studies of first degree relatives of probands with AITD indicated proportions of affected relatives similar to the theoretical expectation for dominant inheritance. More recent family studies have shown that up to 50% of the siblings of patients with the AITDs were TAb positive (23,24), in contrast to 7%–15% in the general population (25). The genetic susceptibility to the production of TAbs was further supported by several segregation analyses in families with Tabs, which also showed a Mendelian dominant pattern of inheritance (26,27). Jaume et al. (28) found evidence for the genetic transmission of TPO antibody fingerprints, also suggesting that autoantibody recognition of the TPO antigen was genetically transmitted. The emergence of knowledge about the human leukocyte antigen (HLA) system and the association between the autoimmune diseases and HLA (see below) solidified the understanding that genetics was important in disease etiology (29). However, our observation showing an association but no linkage to HLA in AITD patients opened up the question of what other genes were involved in disease susceptibility (30).
Gene screening in AITD
There were two choices to develop the genetics of AITD further than thyroid autoantibodies and HLA associations. One was to concentrate on candidate genes and the second was to initiate whole-genome screening. To date, only three groups have reported whole-genome screening data in AITD, but a great many additional reports have been published using association studies of candidate genes (31 –35). In addition to the HLA gene locus, confirmed associations (two or more reports) with AITD include genes for CTLA-4, PTPN22, CD40, CD25, FCRL3, Tg, and the TSHR and there are a significant number of additional gene associations that still await confirmation. We also still await the full results of genome-wide association studies.
Do really significant genes for AITD exist?
Following the clarification that many genes are at work in AITD, the results from whole-genome association studies published for other autoimmune diseases, including type 1 diabetes, lead us to predict that >20 genes may potentially contribute to the AITD phenotypes. However, the question remained whether major genes, those essential to disease development, really exist. If the risk assigned by a particular gene was truly significant, then the responsible gene should be linked to the disease and move with it within families, but we first found that the HLA genes did not obey such a rule for AITD (30), and the search began for what were thought to be the really essential significant genes that caused AITD (36). Really significant genes are considered likely major contributors to genetic risk for a disease. The disease should not occur without some of the responsible polymorphisms. Therefore, such genes should be strongly linked within families and they could become clinically useful as predictors of disease. In the whole-genome screening of families and sibs with AITD, a number of sites were established for GD and HT but none of them had high logatithm (base 10) of odds (LOD) scores, indicating only minor influences (31,32,34). This was found not just for AITD but also for other autoimmune diseases, including type 1 diabetes mellitus. Indeed, even increasing the power of the data by adding more and more families and/or sibs did not help the process. The genes that were linked and/or associated proved to be only minor contributors to genetic risk with low-risk ratios, meaning that every polymorphism was not found in every patient or even the majority of patients, nor was this applicable within all families, and hence the drive to discover more rare single nucleotide polymorphisms (SNPs) on associated genes, by deep sequencing, to see if such linkage can be ascertained by finding more rare polymorphisms (37).
TSHR genetics
The hallmark of GD is the generation of stimulating TSHR autoantibodies and the presence of T cells reactive to TSHR antigen. Hence, the TSHR gene, on chromosome14q31, has long been thought of as a likely disease-specific susceptibility gene. Studies from this laboratory, and those of others, using microsatellite markers were either unable to directly demonstrate significant linkage or association of the TSHR gene with GD, or obtained unimpressive or contradictory data (35,38 –42). Association studies using three common TSHR exonic and nonsynonymous SNPs (in the extracellular and intracellular domain of the receptor) were also inconsistent. However, in two linkage studies (32,39) we described a GD-specific chromosome 14 locus of ∼25 cM, designated GD-1, in the center of which was the TSHR (between markers D14S258 and D14S1054) (Fig. 4). GD-1 was consistently linked with GD and not with HT (31). Subsequently, the study of intronic polymorphisms has been entertained since we now know that intronic DNA may be responsible for regulatory small RNAs as well as providing different start sites for alternative messenger RNA (mRNA) generation. Indeed, we and others (43,44) have also shown previously that the thyroid cell expresses a variety of TSHR mRNA splice variants, indicating that SNPs or small RNAs in this intronic DNA may be important in the generation of different receptor forms and/or their control. A study from Singapore first demonstrated an association of a TSHR intron 1 SNP with GD (45). SNPs in intron 7 of the TSHR were also found to be associated with GD in the Japanese (46), and SNPs in intron 1 of the TSHR were reported to be associated with GD in Caucasians (47). We have recently confirmed this association of intron 1 with GD in Caucasians (48). Using TSHR-SNP-rs2268458, located in intron 1 of the TSHR gene, we genotyped 200 patients with GD, 83 patients with HT, and 118 healthy controls (all female Caucasians). The frequency of the combined genotype (allele) CC + TC was significantly higher in GD patients versus controls, suggesting that the C-containing genotype increased the risk for GD in a dominant manner (p = 0.018, odds ratio [OR] = 1.8). The allele and genotype frequencies from GD patients, but not HT patients, were significantly different from controls. This association of the TSHR with GD has now been reported to be refined to within a 40 kb region of TSHR intron 1 in Caucasian patients with GD (49). Nevertheless, the OR for these TSHR intron 1 SNPs remains low.

Schematic representation of the GD-1 locus on chromosome 14. Using classical linkage techniques and whole-genome screening, we identified a major Graves' disease susceptibility locus on chromosome 14. This locus was named Graves' disease 1 (GD-1). The TSH receptor gene locus is between the sites A and B, from the two studies (32,39). This map also shows that the type 2 iodothyronine deiodinase gene (DIO2) is also very close to the TSHR gene at chromosome 14q31.
Relationship between the TSHR and HLA
HLA-DR3 has been long shown to be associated with GD (29). Recently, the presence of Arg at position 74 of the HLA-DRβ chain has been shown to be strongly associated with GD (50). This finding may provide an attractive mechanism for the association of HLA with GD. T cells recognize and respond to an antigen bound to the HLA molecule expressed on an antigen presenting cell such as a dendritic cell or B cell. It has been well shown that different HLA alleles have different affinities for peptides from an autoantigen (51). Some HLA-DR binding studies have shown higher affinity of HLA-DR3 to TSHR immunodominant peptides, suggesting that certain DR sequences influence the binding and presentation of TSHR peptides (52). This may provide a mechanism by which DRβ 74Arg influences the susceptibility to GD (50) since the presence of HLA-DR3 is a well-established risk factor (53). HLA DR typing was available for ∼80% of the patients in an unselected manner in our recent study, but the analyses showed no evidence for an interaction between TSHR-SNP-rs2268458 and HLA-DR3 in patients with GD, suggesting that each gene was acting independently.
Relationship between the TSHR and the CTLA 4 susceptibility genes
We have examined the interaction between the TSHR gene and the (A/G49) SNP in exon 1 of the CTLA 4 gene, which we and others have previously shown to be associated with AITD (54 –57). There was also no evidence for any additive risk between TSHR-SNP-rs2268458 and CTLA 4 SNP A/G49 in the GD patients. In fact, the OR levels were less than additive in this type of analysis. This suggested that the TSHR gene polymorphism may assign more susceptibility to GD than that assigned by CTLA 4.
Relationship between the TSHR gene and X chromosome inactivation
Biased X chromosome inactivation has also been confirmed by a number of investigators to be greater in patients with AITD (58 –60), but our analyses once again showed that there was no evidence for an interaction between TSHR-SNP-rs2268458 and XCI skewing in GD patients, yet again indicating that the TSHR gene was acting independently. However, such conclusions need to be considered with the knowledge that dependable interaction studies require very large datasets so that lack of gene interaction may be secondary to lack of analytical power.
Possible Mechanisms by Which the TSHR Gene May Influence GD (Table 2)
TSHR splicing
Gene splicing enables a single gene to increase its coding capacity, allowing the synthesis of protein isoforms that are structurally and functionally distinct. At the transcriptional level, tissue-specific mRNAs of 4.3–5.3 kb have been detected as the major species encoding full-length glycoprotein hormone receptors in several eukaryotic systems. However, smaller species incapable of encoding the entire receptor have also been observed. For example, analysis of 80 luteinizing hormone (LH) receptor cDNA clones revealed that 60% encoded full-length LH receptors, whereas the remaining 40% were apparently splicing variants, all of which lacked transmembrane-encoding sequences (61). Similarly, Northern blot analyses have revealed “minor species” of glycoprotein hormone receptor mRNAs that were both less abundant and shorter than the major species (62). One way of forming splicing variants is by the presence of alternative splice sites that may be found within intronic regions. The association of the TSHR gene with GD based on intronic SNPs suggests that such polymorphisms may influence TSHR mRNA splicing. For the human TSHR (hTSHR), the major mRNA species is 4.4 kb, but additional transcripts of 1.7 and 1.3 kb and smaller have been reported by this laboratory and others (43,63). The 4.4 kb species is encoded by all 10 exons. The first 9 exons encode the extracellular domain of the receptor, while exon 10 encodes the TMD and intracellular tail. We have characterized the most abundant hTSHR mRNA variant, a 1.3 kb transcript (called hTSHR v1.3) encoding a TSHR lacking the TMD (43). This variant is co-linear at the 5′ end with the first eight exons present in 4.4 kb hTSHR mRNA and contains at the 3′ end coding and noncoding information not present in the 4.4 kb variant. Another (1.7 kb) variant was not further characterized. The method employed at that time to detect hTSHR mRNA variants required that the species were polyadenylated and shared 5′ coding sequences with the previously sequenced 4.4 kb hTSHR mRNA. Recently, Brand et al. (64) provided preliminary evidence for altered mRNA isoform expression, and the disease-associated genotypes of two SNPs (rs179247 and rs12101255) showed reduced mRNA expression ratios of 4.4 kb TSHR relative to two alternate TSHR mRNA splice variants. However, extrathyroidal expression of TSHR variant 1.3 in thymus (65) would suggest that overt expression of such variants in extra thyroidal tissues may also shape the autoreactive T-cell repertoire in genetically susceptible individuals (66). To date we have not been able to confirm changes in TSHR mRNA ratios, but they remain of potential interest.
No consistent evidence has been found to date for any changes in TSHR residues associated with Graves' disease.
Small RNAs
Another major way by which intronic regions may influence gene action is by the endogenous production of small RNAs, such as microRNAs (miRNAs). miRNAs are small, endogenous, nonprotein-coding, single-stranded segments of regulatory RNAs, ∼18–25 nucleotides in length, which have been associated with the regulation of mRNA expression. So far, we know that they are involved in posttranscriptional control by causing degradation of target transcripts, or by direct translational repression, or by influencing signaling cascades. Recent data also show that some miRNAs can increase translation (67). Therefore, these fascinating molecules may play a large role in gene expression. Since the first miRNA was discovered in Caenorhabditis elegans in 1993 (68), 10,883 entries representing hairpin precursor miRNAs, expressing 10,581 mature miRNA products, have been found in 115 species, including primates, rodents, birds, fish, worms, flies, plants, and even viruses. In humans, 721 miRNAs have been registered in the miRBase database. It is believed that there are over 1000 miRNAs in the human genome and that up to 30%–90% of human genes are regulated by miRNAs. These micromolecules are now known to play important roles in the pathogenesis of human diseases, such as cancer, neurodegenerative and metabolic disorders, diabetes, and viral hepatitis (69 –72). In particular, a single miRNA gene has been demonstrated to have a strong impact on the mammalian immune system (73) having a crucial role in modulating immune responses. It is possible; therefore, that abnormal miRNA expression may contribute to AITD pathogenesis. In this regard, we recently used a computational approach to predict potential regions of TSHR intron 1 that may harbor miRNAs, and identified six candidate regions (C1–C6). Using a programmed series of amplimers, we were able to detect three of these candidates in small RNA preparations from a variety of tissues, including human thyroid, and confirmed them by DNA sequencing (74). These miRNAs may be involved in AITD pathogenesis and deserve careful scrutiny.
The TSHR gene and thyroid neoplasms
Congenital hyperthyroidism is a rare disorder that until recently was considered to be a consequence of maternal GD and transplacental passage of thyroid-stimulating TSHR antibodies (75). However, sporadic cases and a few familial cases of nonautoimmune congenital hyperthyroidism have been reported in which the affected individuals possess activating mutations of the TSHR often, but certainly not exclusively, in exon 10, which encodes the transmembrane region and intracellular tail (76,77). In contrast, somatic TSHR mutations (activating, inactivating, or neutral) are unusually frequent and are found in many thyroid neoplasms—both benign and malignant. There is an increased rate of mutagenesis in the stimulated thyroid gland as evidenced in rats which develop thyroid nodules after prolonged stimulation with TSH (78). The 5th–6th TMDs (TM5–TM6) and the 3rd extracellular loop regions of the TSHR have most of the activating mutations. It is intriguing that the TSHR was thought of as a candidate oncogene for thyroid tumors, although structural studies did not support this view (79). However, Fournes et al. (80) examined the oncogenic potential of a mutant hTSHR or Gsα expressed in rat FRTL-5 cells, and showed that only the mutant TSHR was able to induce neoplastic transformation, as demonstrated by growth in semi-solid medium and tumorigenesis in nude mice. Interestingly, undifferentiated thyroid cancers often have silenced TSHRs. Aberrant methylation of the TSHR promoter in human thyroid carcinomas correlates with lack of, or low, TSHR expression (81). TSHR mRNA from circulating thyroid cancer cells can also be detected in blood of thyroid cancer patients and has been claimed to be a useful molecular marker for thyroid cancer recurrence (82) although the fact that other cells, such as marrow-derived fibrocytes, can also be found in the circulation may compromise this diagnostic approach.
The TSHR gene and longevity
Atzmon et al. (83) reported and confirmed that exceptional longevity is associated with increased serum TSH. Their further study (84) measured serum TSH and free T4 in Ashkenazi Jewish centenarians (n = 232; median age, 97 years), their offspring (n = 366; median age, 69 years), and age-matched controls without familial longevity (n = 163; median age, 70 years). They demonstrated that the offspring of centenarians also have higher serum TSH levels than age-matched controls without familial longevity, and showed a significant but modest degree of heritability for TSH levels in the centenarians and their offspring. Moreover, they found that the allelic frequency of two SNPs (rs12050077 and rs10149689) in the TSHR promoter/enhancer region was associated with the increased serum TSH. The two SNPs showed strong linkage disequilibrium, and their haplotype frequency was significantly higher among centenarians and offspring than among controls. Therefore, carriers with these two SNPs may have higher serum TSH, possibly reflecting mildly decreased thyroid function and longevity. However, the mechanism of this association is unclear. The associated SNPs were located outside the TSHR gene but may be in linkage disequilibrium with a regulatory genetic variant that is located either in the enhancer or in the promoter of the TSHR. Thus, the data demonstrated a possible effect on gene expression that may lead to increased TSH levels, and further studies are needed to clarify such a mechanism. In addition, whether the SNPs inside the TSHR gene are directly associated with longevity also remains to be determined. It remains possible that the elevated level of TSH in centenarians is simply the result of longevity as TSH levels increase with age (83).
The TSHR gene and other abnormalities
Although the TSHR is highly expressed in the thyroid, it is also expressed in brain, bone, fibroblasts, and other sites (85 –87), and mutations in the TSHR have been related to a variety of abnormalities. The TSHR gene has been associated with photoperiod control in birds (88) and polymorphisms in the TSHR gene may be related to the loss of seasonal reproduction cycles in domestic birds (89). The TSHR has been reported to be overexpressed in brains of patients with Downs' syndrome and Alzheimer's disease (90). The TSHR-Asp727Glu polymorphism was associated with reduced serum TSH levels but with increased femoral neck bone mineral density (BMD) (91), and a 50% reduction in TSHR expression in mice generated profound osteoporosis together with focal osteosclerosis (92). A germline mutation of the TSHR gene (P639S) associated with thyrotoxicosis was also associated with mitral valve prolapse in a Chinese family (in three of the four affected patients) (93). Clearly, we have much to learn about these extrathyroidal genetic associations.
Footnotes
Acknowledgments
Supported in part by NIH grants DK069713 and DK052464, and the VA Merit Award Program. We thank Yaron Tomer, M.D., and David Greenberg, Ph.D., for their ongoing collaboration and support.
Disclosure Statement
T.F.D. is a member of the Board of Kronus Inc., Boise, Idaho.
Portions of this review were presented at the Spring 2010 Meeting of the American Thyroid Association, “Thyroid Disorders in the Era of Personalized Medicine,” Minneapolis, MN, May 13–16, 2010.