
Abstract
Background:
Our laboratory identified six distinct inactivating TSHR gene mutations in Arab tribes living in Israel. We recently reported three nucleotide substitutions in exon 3 producing p.[L89L;Q90P] and one in exon 9 of the same allele producing p.P264S in Family A. Family B, reported herein, harbors the identical mutation in exon 3 only. We set to determine whether the mutations have common ancestral origin.
Methods:
Coding regions of the TSHR were sequenced and flanking microsatellite markers spanning 5.3 cM were used for haplotyping.
Results:
Two siblings of Family B were compound heterozygous for TSHR gene mutations. The paternal allele contained the exon 3 mutation and the maternal allele harbored a mutation in exon 10 (p.L653V). We investigated the possibility of a founder effect with subsequent mutational events for the presence of the same exon 3 mutation in different families. The haplotype of the allele harboring the exon 3 mutation in Family B was identical to that of Family A, also harboring the exon 9 mutation on the same allele, indicating that the latter occurred subsequently. The ancestral wild-type TSHR was present in Family B, suggesting that the mutation in exon 3 was also new in the history of that population.
Conclusions:
It is more likely that two consecutive mutational events occurred on the ancestral wild-type allele instead of a recombination bringing exon 3 and exon 9 mutations together on the same allele. New mutational events contribute to the high prevalence of TSHR mutations in this population in addition to a founder effect and limited gene pool due to inbreeding.
Introduction
RTSH was considered to be a rare condition. However, in a recent large population-based Japanese study, the prevalence of monoallelic TSHR mutation was found to be as high as 9.4% in patients found to have high TSH at birth (4). Based on Hardy–Weinberg distribution, the expected frequency of heterozygous TSHR mutation was estimated to be 0.6% in the general Japanese population. Cordella and colleagues found an even higher prevalence of TSHR mutations, 22.9% in a population of young subjects with nonautoimmune subclinical hypothyroidism (5).
From the large population-based investigation in Japan, one particular mutation, c.1349G>A (p.R450H), is due to a founder effect and explains in part the high prevalence of TSHR mutation in Japan (4). Although the occurrence of genetic diseases in an isolated population suggests a founder effect, in many cases the increased frequency is due to more than one mutation (6). Our laboratory has identified six TSHR mutations—c.202C>T (p.P68S), c.267G>T (p.L89L), c.269_270AG>CT (p.Q90P), c.790C>T (p.P264S), c.1825C>T (p.R609×), and c.1957C>G (p.L653V)—in consanguineous Arab Muslim communities living in Northern Israel (7 –10). The frequency of TSHR mutations was 4.3% in one tribe previously not known to have thyroid disease (9). Several hypotheses such as chance phenomenon, migration of families with affected patients, high mutation rates, digenic inheritance, and selective advantages to carriers have been proposed (6).
We previously reported a kindred of 30 individuals from the same Arab population (referred herein as Family A), in which three nucleotide substitutions were identified in exon 3 of the TSHR producing p.[L89L;Q90P] and a mutation in exon 9 of the same allele producing p.P264S (10). Another family of 12 individuals, reported herein (Family B), belonging to a different tribe harbors the identical TSHR mutations in exon 3 but not that in exon 9. The aim of this study was to determine whether the mutations in exon 3 of the TSHR gene in these two families have common ancestral origin.
Materials and Methods
Studies were approved by the Institutional Review Boards of the University of Chicago and written informed consents were obtained.
Index family (Family B)
The propositus (subject III-2) was the second child of a consanguineous Arab Muslim couple, born after an unremarkable pregnancy and labor. This family, originating from Syria, resides in the central Israel. Newborn screening for congenital hypothyroidism revealed TSH of 31.4 mIU/L and a total thyroxine (T4) of 8.5 μg/dL that is below 10th percentile. A serum TSH obtained a few days later was 8.66 mIU/L (normal range, 1.7–9.1) and free T4 (FT4) 12.6 pmol/L (normal range, 11–18.8). At the age of 2 months, he was referred to a pediatric endocrinologist because of hyperthyrotropinemia. Physical examination revealed no sign of hypothyroidism. Laboratory tests found hypothyroidism: TSH 12.7 mIU/L (normal range, 0.35–4.2), FT4 10.6 pmol/L (normal range, 11.4–20.9), and free 3,3′,5-triiodothyronine (FT3) 3.4 pmol/L (normal range, 3.5–6.5). Antibodies to thyroperoxidase (TPO) and thyroglobulin (Tg) were absent. Treatment with 5 μg/kg/day levothyroxine (L-T4) was initiated at the age of 2 months. At the age of 3 years, the thyroid gland had normal size and position that was found by ultrasonography. 99mPertechnetate thyroid scintigraphy showed normal uptake in a eutopic thyroid gland. After discontinuation of L-T4 at 3 years of age, the serum TSH rose to a level of 22.1 mIU/L with a normal FT4 concentration of 12.6 pmol/L. Clinical assessment to date reveals normal growth and development.
His brother (subject III-4) had a similar pattern of thyroid function tests without stigmata of hypothyroidism. He received L-T4 of 5 μg/kg/day since the age of one month and his TSH level was normal. Serum Tg levels before and during L-T4 therapy were 42.2 and 10.7 ng/mL. His growth and development have been normal. Their parents and siblings were clinically and biochemically euthyroid (Fig. 1). The propositus and his brother were found to be compound heterozygotes for TSHR gene mutations. In one allele an AG to CT substitution in exon 3 (c.269_270AG>CT) replaces the normal glutamine at codon 90 with a proline (p.Q90P) and the other allele had a C to G transversion in exon 10 (c.1957C>G) that results in replacement of the normal leucine with a valine at codon 653 (p.L653V). A silent mutation due to a G to T transversion in codon 89 (c.267G>T) that results in no change of leucine (p.L89L) was also found in the allele harboring the exon 3 TSHR gene mutation. These mutations were reported before in Family A (10) and Family C (8,9) (see Family A and Family C below). These findings prompted sequencing of the entire TSHR gene in all family members.
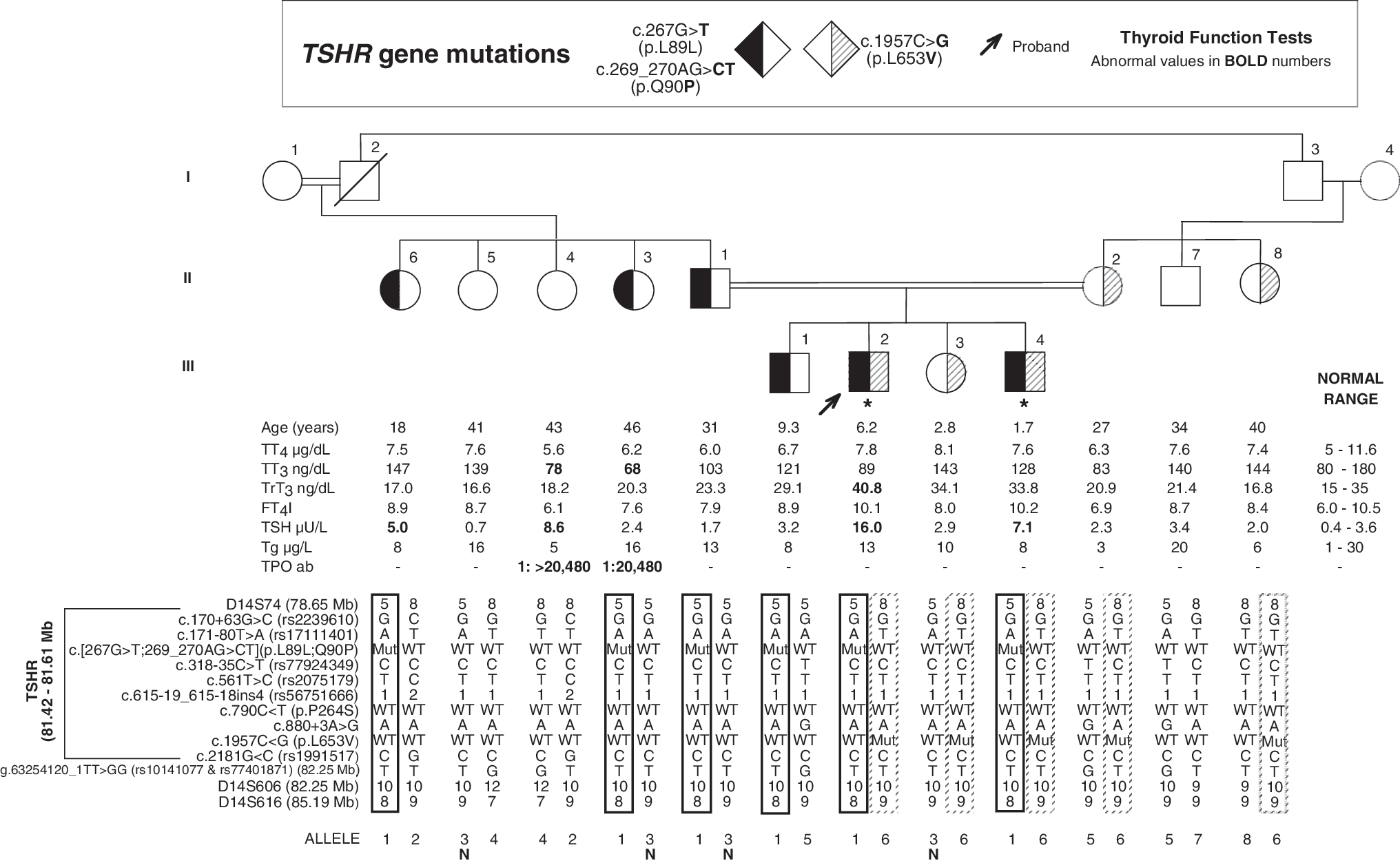
Pedigree and haplotypes of members of the Family B. Square symbols indicate men, circles women, and lozenges any gender. Double lines between spouses indicate consanguinity. The genotype of each subject is indicated within the symbol, as identified in the legend above the pedigree. Results of thyroid function tests are aligned with each symbol and were obtained on no hormonal replacement except for subjects III-2 and III-4 who were on 5 μg L-T4/kg body weight (indicated by asterisk). Roman numerals to the left of the pedigree indicate the generation and numerals on the right of each symbol indicate individual family member. Subjects were tested for the polymorphic markers. The haplotypes are shown below each symbol and outlined with solid or dashed line to trace the inheritance of the different alleles harboring the mutations. The letter N indicates the ancestral allele without mutation.
Family A
Three index cases from an extended kindred of Arab Muslims living in Northern Israel presented with hyperthyrotropinemia or congenital hypothyroidism. We reported this family to have the same three nucleotide substitutions in exon 3 as Family B, but also had a mutation in exon 9 (p.P264S) on the same allele (10).
Family C
The proposita, also from Arab Muslim descent living in Northern Israel, had compensated hypothyroidism. She was homozygous for a missense TSHR gene mutation, p.L653V (8,9). This mutation was presented in one allele of the proband in Family B.
Thyroid function tests
Total T4, total T3, and TSH were measured by chemiluminescence immunometric assays using the Elecsys Automated System (Roche Diagnostics, Indianapolis, IN). Total rT3 (reverse T3 or 3,3,5′-triiodothyronine) was measured by a commercial RIA (Adaltis Italia, Bologna, Italy) and Tg by an in-house RIA (11). The FT4 index and FT3 index were calculated as the product of the total serum concentrations of each iodothyronine and the normalized resin T4 uptake ratio. Antibodies against Tg and TPO were measured by passive hemagglutination (Fujirebio, Inc., Tokyo, Japan).
Molecular analysis
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit (QIAGEN, Valencia, CA) according to the manufacturer's instruction. For sequencing, all 10 coding regions and flanking introns of the TSHR gene were amplified using primers and thermocycler setting similar to those previously described (12). The resulting polymerase chain reaction products were visualized on 1.8% agarose gel before direct automated sequencing.
Several polymorphic markers in exonic and intronic regions of the TSHR gene (rs2239610, rs17111401, rs77924349, rs2075179, rs56751666, and rs1991517) were used for haplotyping. Three standard polymorphic microsatellite markers (D14S74, D14S606, and D14S616) spanning about 6.54 Mb or 5.3 cM (Marshfield map) around the TSHR locus were also used for haplotyping as detailed previously (13). The rs10141077 and rs77401871, single-nucleotide polymorphisms (SNPs) that were identified during direct sequencing of D14S606, were included for analysis. Nucleotide numbering for mutations and variants uses cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference human sequence, NM_000369.2.
Statistical analyses
Data of thyroid function tests expressed as mean±SD were analyzed by two-tailed Student's t-test for unpaired observations.
Results
Genetic and clinical results
Two previously described loss-of-function mutations of TSHR gene, p.Q90P and p.L653V, were identified in the propositus on different alleles. In addition, a silent mutation p.L89L was found on the same allele as the p.Q90P mutant allele and identical to that of Family A reported previously (10). Of the 12 family members studied, 9 were found to harbor TSHR gene mutations (Fig. 1). The propositus and his brother (subjects III-2 and III-4) were compound heterozygous for p.[L89L;Q90P], inherited from father, and p.L653V, inherited from mother. Father, two paternal aunts, and oldest brother of the propositus (subjects II-1, II-3, II-6, and III-1) were heterozygous for p.[L89L;Q90P]. Mother, maternal aunt, and sister of the propositus (subjects II-2, II-8, and III-3) were heterozygous for p.L653V.
The intronic variant c.880+3A>G identified in the alleles of subjects II-2, II-7, and III-1 was presumed not to produce exon skipping. A guanine is found at position +3 in 35% of intron donor sites, known as consensus sequence (14). Also heterozygotes for this variant had normal serum TSH concentration.
The serum TSH concentrations of subjects heterozygous for the TSHR gene mutations p.L89L;Q90P (n=4, TSH 3.08±1.42 mIU/L) and heterozygous for p.L653V (n=3, TSH 2.4±0.45 mIU/L) were not statistically different than the TSH concentrations of family members with wild-type (WT) alleles (n=3, TSH 4.23±4.02 mIU/L). Three family members with TSHR gene mutations were receiving L-T4 treatment: two (III-2 and III-4) were compound heterozygous and one (II-3) was heterozygous for p.[L89L;Q90P] and had anti-Tg antibodies.
Haplotype analysis
Analysis of microsatellite markers (D14S74, D14S606, and D14S616) and two SNPs (rs10141077 and rs77401871) spanning 5.3 cM of the TSHR locus, known intragenic polymorphic markers (rs2239610, rs17111401, rs77924349, rs2075179, rs56751666, and rs1991517), and a variant of intron 9 donor splice site (c.880+3A>G) residing in TSHR gene generated a common haplotype among individuals having p.[L89L;Q90P] allele in Family B. In subjects II-1 and II-3 heterozygous for p.[L89L;Q90P], the WT allele had the same haplotype (allele N, see Fig. 1) except for the exon 3 mutation and D14S616 located 3.66 Mb downstream from the exon 3 mutation site. This later difference could be from recombination or mutation because microsatellite markers have higher mutation rates than SNPs. Furthermore, subjects from Family A, with exon 3 and exon 9 mutations on the same allele, had an identical allele as subjects from Family B with exon 3 mutation, except for the exon 9 mutation and D14S616. These haplotypes support the notion that exon 3 and exon 9 mutations developed on this ancestral WT TSHR allele identified in Family B (Table 1) through consecutive mutational events. In addition there is evidence of common ancestry for the allele harboring the p.L653V mutation of Family B with the allele harboring the same mutation in Family C as they share identical haplotypes except for marker D14S606 (Table 1).
Previous reported TSHR mutations in Arab population, c.202C>T (p.P68S) and c.1825C>T (p.R609×), were not found in these families.
Marker location on the reference human sequence (NM_000369.2).
Ex, exon;
Discussion
Data on the prevalence of inactivating mutations in the TSHR gene in patients with congenital hyperthyroidism or nonautoimmune hyperthyrotropinemia are variable, from 2% to 40% depending on selection criteria of the population studied (9,15 –22). Population-based genetic study in Japan showed that the TSHR mutations are not an uncommon cause of RTSH (4). However, a founder mutation that accounts for about 70% of mutants is responsible for the high prevalence of TSHR mutations in this population.
To date, six inactivating mutations have been found in Israeli Arab Muslim patients with congenital hypothyroidism belonging to three large extended kindreds (7,9,10). With the exception of p.R609×, these mutations were found only in this population. The p.R609× mutation was also reported in a consanguineous family of Turkish origin (23). The combination of common ancestry and intense cultural consanguinity accounts for a high inbreeding rate in these populations. Our previous study showed a prevalence of 4% in a control population of the same origin with no known thyroid diseases (9). In the current study, we demonstrated founder effect for the p.[L89L; Q90P] and p.L653V mutations.
Furthermore, two consecutive mutational events were demonstrated by haplotyping of the TSHR locus. This supports the hypothesis that the high prevalence of TSHR mutations in this Arab Muslim population is caused not only by a founder effect and limited gene pool due to breeding, but also by new mutation events. From the haplotyping study, we identified a specific allele carrying the mutants (except for the difference in the distal markers that could result from recombination after several generations). The allele with the exon 3 mutation (p.[L89L;Q90P]) was found to be identical to an ancient WT TSHR allele also present in Family B with all genetic markers on these alleles being identical, except for the exon 3 mutation. Moreover, our data suggest that the exon 9 (p.P264S) mutation occurred on this already mutated allele to generate the allele present in Family A that harbors both exon 3 and exon 9 mutations. This is supported by the fact that the other genetic markers on these alleles, including the distal markers rs1991517, rs10141077, rs77401871, and D14S606, are identical and not different as it would be expected if the mutation p.P264S occurred by recombination. The diversity of WT allele haplotypes strengthens the hypothesis of common ancestral allele.
Many different inactivating TSHR gene mutations including missense, nonsense, frameshift deletions/insertions, and splice site mutations have been documented to be responsible for RTSH. These loss-of-function mutations are found throughout the receptor sequence, without clear localization to hot spots such as CpG regions (24). Whether mutations in the TSHR gene may impart a selective advantage to carrier subjects (6,25) will require more studies to explore this hypothesis.
Mean TSH concentrations in the subjects with monoallelic inactivating TSHR mutations, p.[L89L;Q90P] and p.L653V, and with the WT TSHR were not significantly different. However, the sample size is too small to draw a definite conclusion. Additional family members refused to participate in the study. The subjects compound heterozygous for the two mutations had compensated hypothyroidism. These results are compatible with high residual function of the mutant TSHRs as shown by functional analyses in the previous studies (9,10). This indicates that these two mutations have a mild effect on TSHR inactivation when present in heterozygotes or even together in compound heterozygotes. Our previous study showed that heterozygotes for the p.L653V allele had significant higher mean TSH level than family members with WT allele only (9). Thyroid function tests can vary between different individuals affected with the same mutation or among members of the same family. These findings suggest that environmental factors (e.g., thyroid hormone requirement, iodide supply, or acquired thyroid insults) as well as yet unknown genetic modifiers may account for this difference in the phenotype.
In conclusion, we propose that two consecutive mutational events occurred on an ancient WT allele generating the allele with both exon 3 and exon 9 mutations, instead of a recombination event that brought exon 3 and exon 9 mutations together on the same allele. This indicates that new mutational events contribute to the high prevalence of TSHR mutations in this population in addition to a founder effect and limited gene pool due to inbreeding.
Footnotes
Acknowledgment
The project described was supported in part by grants R37DK15070, T32DK007011, P60DK20595 from the National Institute of Diabetes and Digestive and Kidney Diseases and 5M01RR04999. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health.
Author Disclosure Statement
S.R. consults for Quest Diagnostics. The remaining authors have no competing financial interests.