
Abstract
Background:
Thyroid dysfunction is a well-known adverse effect of sunitinib, a drug that targets multiple receptor tyrosine kinases, including vascular endothelial growth factor receptor (VEGFR). As several kinds of tyrosine kinase inhibitors (TKIs) are now available, this has been postulated to be a side effect of the TKIs that target the VEGFR (VEGF-TKIs). However, sunitinib, one of the first-generation TKIs, likely causes thyroid dysfunction more frequently than other TKI classes, leading not only to hypothyroidism, but also to thyrotoxicosis.
Summary:
Based on the reports published to date, including our own studies, we have hypothesized that sunitinib may exert these effects, because it targets a broad spectrum of tyrosine kinases. This not only includes VEGFR2, but also VEGFR1 and the platelet-derived growth factor receptor (PDGFR). This, in turn, may suggest that not only VEGFR2 but also the PDGFR and/or the VEGFR1 play an important role during angiogenesis in the thyroid.
Conclusions:
Our current hypothesis may explain the mechanisms that underlie TKI-induced thyroid disorders. By learning how various kinds of TKIs affect thyroid function, we may elucidate how the angiogenesis in thyroid is regulated both physiologically and pathologically.
Introduction
Review
Clinical studies of sunitinib
Sunitinib is a multitarget TKI with activity against VEGFRs, platelet-derived growth factor receptor (PDGFR), KIT, and RET. In several countries, including the United States, Europe, and Japan, this drug is approved for the treatment of metastatic renal cell cancer (RCC), imatinib-resistant gastrointestinal stromal tumors (GISTs), and highly differentiated unresectable pancreas neuroendocrine tumors. A possible relationship between sunitinib therapy and thyroid disorder was first noted in a phase I/II clinical study for patients with imatinib-resistant GISTs. Two index-case patients in this trial showed typical signs and symptoms of hypothyroidism such as fatigue, confusion, cold intolerance, hoarseness, and constipation (4). Before this clinical study, fatigue had been attributed to a direct effect of sunitinib (5).
These cases led to a prospective, observational cohort study designed to better describe the prevalence and clinical presentation of thyroid dysfunction related to sunitinib therapy. Forty-two patients who received sunitinib for at least three cycles and who had normal baseline serum thyrotropin (TSH) values were observed for a median of 37 weeks (4). Abnormal serum TSH concentrations were noted in 26 of these patients (62%). Fifteen patients (36%) developed persistent primary hypothyroidism; 4 (10%) developed isolated TSH suppression; and 7 (17%) showed transient TSH elevations. Among the patients with hypothyroidism, 15 (40%) had a suppressed TSH before developing hypothyroidism, a constellation suggestive for thyroiditis.
Since these observations, thyroid dysfunction associated with sunitinib gained considerable attention and was the subject of many cohort studies (4,6 –30), although the patient numbers were never large (Table 1). Retrospective studies have indicated that sunitinib can induce hypothyroidism, including subclinical hypothyroidism with TSH levels above the reference range and peripheral thyroid hormone levels within the normal range in 53%–85% of patients (6,7). In prospective studies, this complication has been reported in 27%–71% of patients (4,8 –14). In contrast to the situation with interferon and imatinib, the incidence and severity of hypothyroidism are reported to be unassociated with the prior treatment, nor with the existence of thyroglobulin antibodies, but it is associated with the duration of therapy (4,6).
Patients with (sub)clinical hypothyroidism (SC HT), including patients who showed transient elevation of TSH.
Whether hypothyroidism may serve as a predictive marker of treatment outcome or not: +, yes; −, no; Ø, not reported.
VEGF-TKIs, tyrosine kinase inhibitors that target vascular endothelial growth factor receptor (VEGFR); pts, patients; TSH, thyrotropin; GIST, gastrointestinal stromal tumors; RCC, renal cell carcinoma; meso, mesothelioma; HCC, hepatocellular carcinoma; NSCLC, non–small-cell lung cancer; DTC, differentiated thyroid cancer; MTC, medullary thyroid cancer; Pro, prospective; Retro, retrospective; Ret/Pro, initially retrospective followed by prospective evaluation; ND, not determined; TPO; thyroid peroxidase.
Angiogenesis in the thyroid gland and mechanism underlying sunitinib-induced thyroid dysfunction
The pathogenesis of sunitinib-induced thyroid dysfunction has remained unknown to date, although several mechanisms have been proposed (31). It has been reported previously that 6 of 15 (40%) patients showed a suppressed TSH before developing hypothyroidism, and that two patients with hypothyroidism evaluated by ultrasonography had no evident thyroid tissue despite initially presenting with a normal baseline thyroid function (4). Hence, sunitinib-induced hypothyroidism seemed to be caused by destructive thyroiditis (4). We have also evaluated a number of severely affected patients with hypothyroidism who showed an atrophic thyroid after destructive thyroiditis. It is now known, however, that the frequency of a suppressed TSH is far lower than the incidence of hypothyroidism without a history of a suppressed TSH in patients treated with sunitinib.
The elucidation of the cause of sunitinib-induced hypothyroidism, sometimes followed by destructive thyroiditis, was thus another important question. Based on the fact that the thyroid gland is well vascularized and shows the highest blood flow rates per unit weight of any tissue in the body (32), we attribute sunitinib-induced thyroid disorder largely to its antiangiogenic effects via inhibition of the VEGFR signaling pathway. Consistently, it has been reported that the thyroid blood flow is mainly dependent on the VEGFR signaling pathway in both the normal thyroid gland and in highly vascularized thyroids of patients with Graves' disease. TSH and the IgG present in patients with Graves' disease upregulate the expression levels of mediators such as VEGF-A through the stimulation of the TSH receptor on thyroid follicular cells, from which those mediators are secreted. The secreted VEGF-A then stimulates the VEGFR expressed on endothelial cells in a paracrine manner, and the stimulated endothelial cells start to proliferate and fuse to each other with increased levels of VEGFR1 and VEGFFR2, so that the vascular lumen increases in size (Fig. 1) (33 –36).
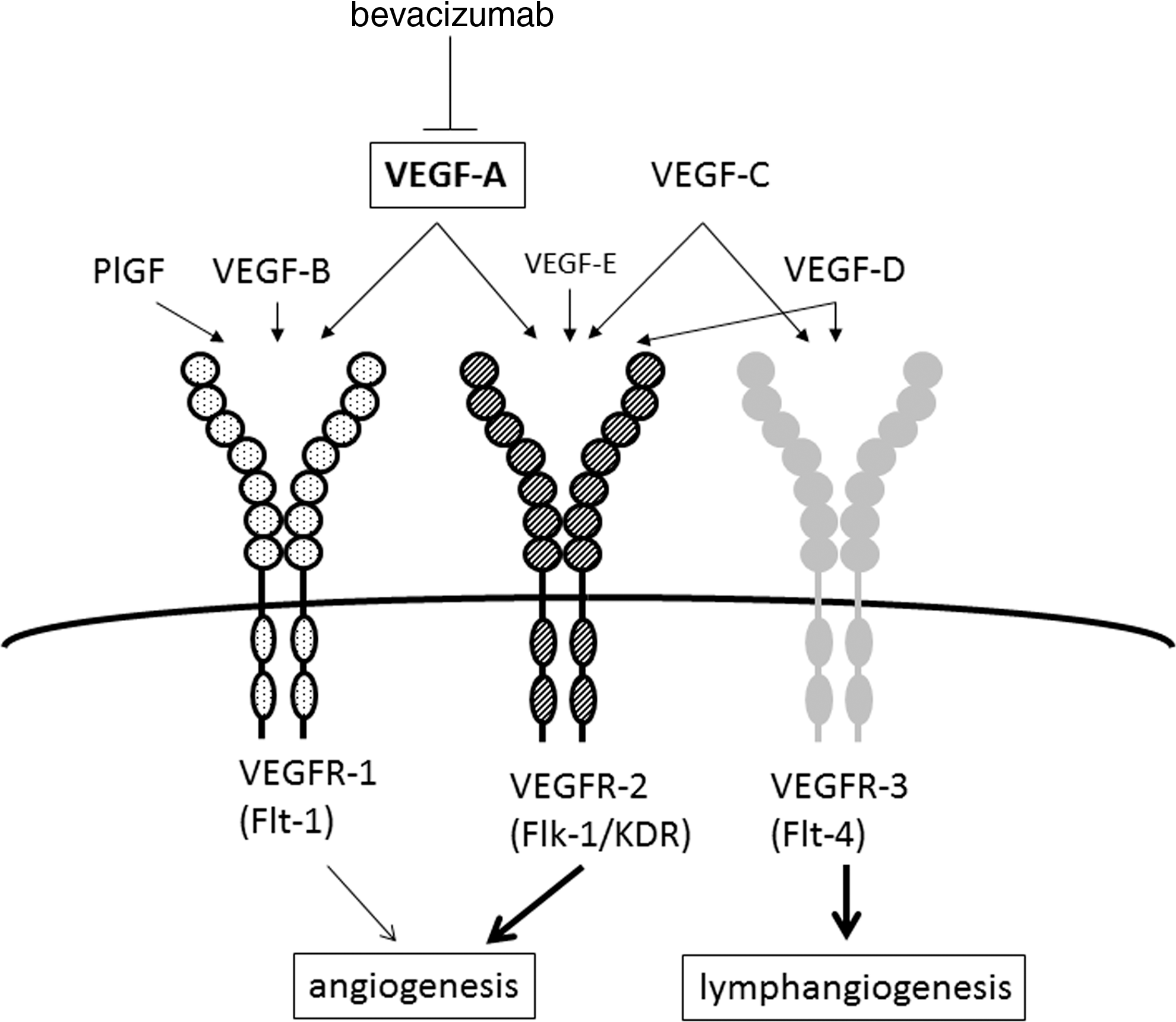
Roles of vascular endothelial growth factor (VEGF) family members, and their receptors. VEGFR1 signaling in addition to VEGFR2 signaling regulates angiogenesis, while VEGFR3 signaling plays a role in lymphangiogenesis. The VEGFR1 is not only activated by VEGF-A but also by placenta growth factor.
Previously, we published a case report providing evidence that sunitinib induces hypothyroidism by reducing blood flow via capillary regression and constriction (37). The patient in question displayed overt hypothyroidism with an atrophic thyroid and a marked reduction in vascularity, as determined by ultrasonography and despite high levels of TSH, one week after sunitinib was commenced. In contrast, during the off-periods in the sunitinib treatment cycles for this case, the volume of her thyroid recovered with an increase in vascularity despite a low level of TSH. These results suggested that thyroid function and volume may depend on its vascularity, which is negatively regulated by sunitinib. One in vivo study supports this hypothesis: rats exposed to sunitinib for 8 days show decreases in microvessels, counted by the total number of thyroid vessels and vessel-to-follicle ratio on histological examination, and recover when sunitinib is withdrawn (38). The same authors previously showed that the administration of sunitinib is associated with an increase in the plasma endothelin-1 concentrations (39). They also showed that the decrease in microvessels in rats exposed to sunitinib and an endothelin receptor blocker was less pronounced compared with rats exposed to sunitinib only. They speculated that, in addition to structural rarefaction (a reduction in capillary density), functional rarefaction (a decrease in perfused microvessels) contributes to the decrease in thyroidal microvessels observed during sunitinib exposure.
It may be reasonable to postulate that when ischemia is too severe for thyrocytes to survive, apoptosis ensues, causing destructive thyroiditis in the absence of any autoimmune disorder (37). This suggestion is compatible with the findings of many reports demonstrating that sunitinib-induced thyroid dysfunction does not seem to correlate with the autoimmune processes responsible for the thyroiditis caused by interferon-alpha or ipilimumab, a monoclonal antibody against cytotoxic T-lymphocyte antigen 4 (40). While hypothyroidism associated with thyroid atrophy following destructive thyroiditis had been reported (4,10), it was recently shown also that sunitinib or sorafenib causes an apparent thyroid size reduction in patients with pre-existing hypothyroidism (41). This reduction is not surprising if sunitinib causes both structural and functional rarefaction of microvessels. Although impaired uptake of radiolabeled iodine during sunitinib treatment has been reported (8), sunitinib did not inhibit iodide uptake in cultured rat FRTL-5 thyroid cells in another in vitro study (42). This discrepancy likely reflects the decreased number of structural and functional capillaries necessary for the delivery of iodide to the thyroid (38).
We hypothesize from the available evidence that sunitinib may reduce the thyroid micropapillary and vascular lumen through the inhibition of basal activity of the VEGFR signaling pathway in a normal thyroid and thereby cause tissue ischemia via reduced blood flow, leading to thyroid dysfunction (6,37,43).
Why does sunitinib induce thyroid dysfunction at a particularly high frequency? A hint from the inhibition of the VEGFR signaling pathway by bevacizumab
If our hypothesis is valid, the TKIs not targeting VEGFRs would not induce thyroid dysfunction. There are few reports to date showing that such TKIs induce thyroid dysfunction in patients with a normal thyroid gland (44,45), whereas imatinib affects thyroid function by inducing the metabolism of levothyroxine in athyroid patients (46). We evaluated the findings available to date for a recombinant monoclonal antibody against VEGF-A, bevacizumab. By preventing VEGF-A from accessing its receptor, bevacizumab inhibits the downstream angiogenesis that is amplified in some cancers, and it has been approved for the treatment of metastatic colorectal cancer and non–small-cell lung cancer, as well as metastatic renal cell carcinoma. Instructively, there are few reports showing that bevacizumab affects thyroid function (47). To further assess the fundamental mechanisms involved, it is necessary to review the basics of the VEGFR signaling pathway.
In humans and other mammals, the VEGF family consists of six related glycoproteins, VEGF-A to -E and placenta growth factor (PlGF; Fig. 1). On the other hand, there are three VEGF receptors (VEGFR1–3). VEGFR1 and VEGFR2 bind VEGF-A and play a central role in the regulation of angiogenesis, whereas VEGFR3 tightly binds VEGF-C and -D and stimulates lymphangiogenesis. Although VEGFR1 binds PlGF and VEGF-B, VEGF-A and VEGFR2 play a key role in angiogenesis, because the tyrosine kinase activity of VEGFR1 is one order-of-magnitude weaker than that of VEGFR2. This may be why the strategy of trapping VEGF-A is more effective. However, it has been reported that PlGF is expressed at a low level under normal physiological conditions in several tissues, such as the heart, lung, skeletal muscle, and thyroid (48), and that in the normal human thyroid gland, PlGF, in addition to VEGFs, is a paracrine mediator produced by thyrocytes (35). Although the angiogenic activity of PlGF and VEGF-B is usually about 10-fold weaker than that of VEGF-A, due to their limited accessibility to only VEGFR1 (49), it is possible that PlGF may play a residual role in normal angiogenesis in the thyroid. In addition, under pathological conditions such as tumor growth, or heart, limb, and ocular ischemia, the knockout of the PlGF gene resulted in impaired angiogenesis, although knockout studies show that PlGF is redundant for vascular development (50 –52).
In vitro studies have indicated that PlGF and VEGFR1 are upregulated in cells exposed to hypoxia (53,54). Clinically, it has been reported that antiangiogenic treatment increases circulating PlGF (55 –58). In thyrocytes, analogously, bevacizumab may upregulate the expression of PlGF via ischemic stress by trapping VEGF-A. Not only under physiological conditions, but also in pathological conditions after treatment with bevacizumab, PlGF may play a hidden role in maintaining the vascularity of the thyroid. At least in part, this possibility would explain why sorafenib, which does not target VEGFR1, causes thyroid dysfunction less frequently (15,16), with the exception of a clinical study in Japan (17) (Table 1).
Insights from other VEGF-TKIs
From the evidence presented in the discussion above, we suspected that not only VEGFR2 but also VEGFR1 may need to be inhibited to cause thyroid ischemia. If efficient inhibition of the VEGFR pathway were sufficient to induce ischemia in the thyroid gland, more potent and specific TKIs that target the VEGFRs would tend to cause thyroid dysfunction more frequently. However, as outlined below, the selective VEGFR inhibitors, axitinib and tivozanib, do not cause hypothyroidism at a frequency as high as sunitinib.
The third-generation TKI, axitinib, which was recently approved in the United States for treating advanced RCC, is a highly selective inhibitor of the three VEGFRs and has a lower potency for inhibiting PDGFR and c-KIT (59). This selectivity is evidenced by the half-maximal inhibitory concentration (IC50) for axitinib, which is 10-fold lower for the VEGFRs than for other family receptors, such as PDGFR. The IC50 values for axitinib against most of the targets of interest are also lower than that of other TKIs, indicating its potency (Table 2) (59 –68). In a phase III trial in the United States (16), which compared axitinib with sorafenib for advanced RCC, 69 of 359 patients (19%) displayed hypothyroidism in the axitinib group, compared with 29 of 355 patients (8%) in the sorafenib group (Table 1). However, only one patient (<1%) showed grade-3 adverse effects (representing symptoms interfering with activity of daily living and resulting in hospitalization), and no grade-4 effects (representing life-threatening myxedema coma) arose in any patient. In clinical trials in Japan (18,19), however, axitinib-induced thyroid dysfunction was reported to occur more frequently, although no cases of grade 3 or 4 hypothyroidism were described (Table 1). Considering the results of clinical trials with sorafenib (17) and linifanib (20), the results from Japan might suggest that Japanese are more susceptible to thyroid ischemia as a result of TKI therapies. While the underlying mechanism remains unclear, we suspect that there may be a correlation with nutritional iodine intake. Similar to axitinib, a third-generation TKI, tivozanib, is also a selective and potent inhibitor of all three VEGFRs. In a phase II trial of this drug, no cases of hypothyroidism were reported (69).
50%
IC50 values are reported in nanomoles. Boldface indicates that sunitinib shows a higher affinity for both VEGFR1 and PDGFR than VEGFR2.
Sources: aSchmidinger and Bellmunt (63); bWilhelm et al. (64); cHu-Lowe et al. (59); dEscudier and Gore (60); eNakamura et al. (65); fAlbert et al. (66); gBhide et al. (67); hWilhelm et al. (68); iPolverino et al. (61); jWedge et al. (62)
Converted from value obtained in the presence of 2.3% bovine serum albumin.
VEGFR, vascular endothelial growth factor receptor; PDGFR, platelet-derived growth factor receptor.
The possible role of non-VEGF signaling pathways in thyroid angiogenesis warrants scrutiny and discussion. Under physiological conditions, it has been reported that thyroid angiogenesis is regulated mainly by VEGF signaling, but that non-VEGF signaling pathways also regulate angiogenesis in the thyroid gland (35,36). It is therefore not surprising if under pathological condition such as ischemia and hypoxia, non-VEGF angiogenic factors such as PDGF and their receptors are secondarily upregulated in the thyroid gland in a compensatory manner. We hypothesize that a higher affinity for both VEGFR1 and PDGFR than VEGFR2 may play a key role in the high frequency of sunitinib-induced thyroid dysfunction; this is called the relative affinity hypothesis. The relative affinities of VEGFR1 to VEGFR2 of sunitinib, axitinib and tivozanib are 0.2, 0.5, and 0.9, while those of PDGFR to VEGFR2 are 0.8, 8, and 1.1, respectively (Table 2).
Several VEGF-TKIs are now approved or under clinical trial. Unfortunately, however, since the methods of evaluating thyroid dysfunction and of displaying it as an adverse effect are completely different between each clinical trial (in addition to differences in patient number, background, and ethnicity), we cannot simply compare the results. For example, the reported frequency of cediranib-induced hypothyroidism is quite different between trials (21,22) (Table 1). In terms of other VEGF-TKIs besides cediranib, as shown in Table 1, the frequency of hypothyroidism tends to be higher in trials in Japan. In spite of these data, clinical trials in patients retaining their thyroids seem to support our hypothesis. For example, Table 1 shows a low incidence of hypothyroidism in patients treated with pazopanib (23), vatalanib (24), linifanib (25,26), and motesanib (27). This is consistent with our relative affinity hypothesis in the point that each VEGF-TKI targets PDGFR with less affinity than VEGFR2: the relative affinities of PDGFR to VEGFR2 of pazopanib, vatalanib, linifanib, and motesanib are 1.8, 15.6, 3.1, and 28, respectively (Table 2).
These observations might support our hypothesis that among several VEGF-TKIs, the molecules that target both the PDGFR and VEGFR1 with a higher affinity than VEGFR2 will frequently induce thyroid dysfunction (Table 2). This may be important to sufficiently block residual signals of thyroid angiogenesis, because the clinically prescribed dose of each TKI is mainly chosen based on its inhibitory effects against VEGFR2 signaling. By knowing the types of kinases that each TKI targets, and its respective selectivity and potency, we will be able to approximately predict whether it might more frequently induce thyroid dysfunction. Hence, by learning how new TKIs affect thyroid function in clinical settings in the future, the physiological and pathological basis for how thyroid angiogenesis is regulated will be further revealed. The potential role of PlGF or PDGFR, as discussed above, could also be properly assessed if we evaluate thyroid function in patients or in animal models using PDGF or PlGF antibodies, in addition to a VEGF-A antibody.
Possible additional mechanisms underlying hypothyroidism induction by TKIs
A previous report has shown that athyroid patients who are on levothyroxine replacement need an increased amount of levothyroxine during sunitinib administration (70). This suggests an additional action of sunitinib on nonthyroidal targets. Also, it was reported recently that in sunitinib-treated rats the serum T4 and T3 decreased, accompanied by increased hepatic type 3 deiodinase (DIO3) activity, decreased type 1 (DIO1) deiodinase activity, and a thyroid histology that showed marked capillary regression (38). In fact, the authors showed prospectively in patients that the T3/reverse T3 ratio decreased during treatment with sunitinib, accompanied by a twofold increase in the TSH levels. Based on these results, they proposed a metabolic hypothesis in addition to a capillary regression hypothesis as the mechanisms of sunitinib-induced hypothyroidism (38). It has been reported in this regard that hypoxia induces expression of the DIO3 gene via a hypoxia-inducible factor (HIF)–dependent pathway (71). Hence, it may be that sunitinib secondarily induces the expression of HIF-1 through its antiangiogenic effects and increases the expression of the DIO3 gene in peripheral tissues such as the liver, where the angiogenic signaling pathways might be different from those in the thyroid.
If this is the case, we may postulate that even metabolic effects may be explained by the antiangiogenic effects of TKIs, although the target organs are different. The finding that patients on sorafenib showed decreased T3/T4 and T3/rT3 ratios, accompanied by decreased serum thyroid hormone levels and increased TSH levels (72), is compatible with the concept of a VEGF-TKI class effect. In clinical trials of motesanib and vandetanib in thyroidectomized patients, both of these TKIs frequently caused substantial increases in the levothyroxine requirement. As with sunitinib, both drugs might cause an increased metabolism of levothyroxine via an increased activity of D3 in peripheral tissue as a TKI class effect (28 –30). In contrast, in patients who retain their thyroid gland, neither drug induced hypothyroidism frequently (Table 1) (27,73). These results are also compatible with our hypothesis concerning thyroid angiogenesis, given the fact that motesanib targets PDGFR with far less affinity than the VEGFR2, and vandetanib targets neither VEGFR1 nor PDGFR (Table 2) (61,62).
When hypothyroidism develops under therapy with TKIs, TSH levels are often inappropriately high for the concomitantly measured serum free T3 and free T4 levels. The marked increase in TSH levels suggests a potential interference of TKIs on thyroid hormone action at the pituitary or hypothalamus level. If the activity of type 2 deiodinase (DIO2, a 5′-deiodinase expressed in the pituitary gland), decreases far more than that of DIO1 (expressed mainly in the liver and kidney) while on sunitinib therapy, it would lead to the intracellular depletion of T3 in the thyrotrophs and an inappropriately high TSH level. In terms of the intracellular depletion of T3, another group has referred to the possibility that TKIs might interfere with the monocarboxylate transporter 8 (MCT8), which is a thyroid hormone transmembrane transporter in the brain, including the hypothalamus and pituitary, and also in the liver, kidney, and other tissues. These authors showed that several TKIs, including sunitinib and imatinib, dose dependently inhibit MCT8-dependent T3 and T4 uptake in a noncompetitive manner in vitro (74). Based on the molecular structures, the authors speculate that TKIs might fit into the iodothyronine-binding sites of transmembrane transporters. If this happens in vivo, a therapeutic dose of a TKI might cause inhibition of the negative feedback role of T3 at the pituitary and the hypothalamus, and lead to an inappropriately high TSH level.
Sunitinib-induced thyroid dysfunction and cancer treatment outcomes
Thus far, we have proposed that thyroid ischemia via capillary regression and constriction is most likely due to the inhibition of VEGFR1 and 2 and PDGFR as the underpinning mechanism of sunitinib-induced thyroid dysfunction. If this hypothesis is correct, patients developing thyroid dysfunction might tend to have their cancer controlled better through the common antiangiogenic effects affecting both the tumor and the thyroid. Since sunitinib-induced hypothyroidism has gained attention, the possibility that hypothyroidism can be a surrogate marker for the efficacy of sunitinib has been discussed (75). There are several studies (11 –13) that report that patients developing hypothyroidism, some of whom were supplemented with levothyroxine, had a significantly better prognosis (Table 1). This suggests that the effectiveness of sunitinib might depend, at least in part, on its efficacy against TKs such as VEGFRs and PDGFR expressed in vascular endothelial cells in the host, in addition to the properties of the tumor. However, another study could not find any difference in progression-free survival between patients who showed thyroid dysfunction, followed with hormonal substitution, and patients who did not (14). Larger comparative studies are thus needed to validate these aspects.
Summary and Conclusion
In this review, we propose that sunitinib induces thyroid dysfunction more frequently than other TKIs, because it targets a broad spectrum of tyrosine kinases, that is, inhibiting not only VEGFR2 but also PDGFR and VEGFR1 more potently, thereby inducing thyroid ischemia via capillary regression and constriction. This may, in turn, suggest that not only VEGFR2 but also other kinases such as PDGFR and VEGFR1 are likely to play an important role in angiogenesis in the thyroid. In addition to their antiangiogenic mechanism, acceleration of thyroid hormone metabolism through increased DIO3 activity by TKIs may result in an additional effect on thyroid function, although this may also be explained by the antiangiogenic effects of TKIs. In the future, by learning how new TKIs affect thyroid function, we may be able to elucidate how angiogenesis in the thyroid is regulated both physiologically and pathologically. Although thyroid dysfunction as a result of TKI therapy is manageable, and these drugs can continue to be used due to their clinical benefits, it may be useful to predict how severely thyroid function may be affected by new TKIs, so that clinicians can detect and manage any thyroid disorders at an early stage of the treatment.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.