
Abstract
Background:
Synovial sarcomas are uncommon malignancies that mainly affect adolescents and young adults. Most arise from the deep soft tissues of the extremities, but they can occur in other parts of the body such as the lung. Synovial sarcomas after radiation therapy are rare, in contrast with other sarcomas, with only six reported cases. Secondary malignancies after radioactive iodine (RAI) therapy are also uncommon, with the most consistent evidence for hematologic malignancies.
Patient Findings:
We present what we believe to be the first report of a synovial sarcoma of the lung with an SS18/SSX1 translocation after RAI therapy. At age 20, the patient developed papillary thyroid cancer and later had two surgically confirmed recurrences. Over the course of her care, she received a total of about 220 mCi of RAI. At age 34, as part of an evaluation for another suspected recurrence, she had a position emission spectroscopy-computed tomography scan, and a pulmonary mass was detected.
Summary and Conclusion:
Although not previously reported, this case suggests that synovial sarcomas may be a secondary malignancy after RAI therapy. The latency in this case is reasonable, the dose to the lungs was small, but in the range where radiation-related malignancy may occur, and the somatic chromosomal rearrangement could be a radiation effect.
Introduction
We describe a woman who developed a rare tumor, a synovial sarcoma of the lung after receiving RAI therapy for a recurrent papillary thyroid cancer. Although some cases of secondary primary lung malignancies have been described (3,4), Sawka et al. did not observe a statistically significant increase in them (2).
Patient
The patient was 20 years old in 1997 when she was diagnosed with a papillary thyroid carcinoma. She has no children, and there is no history of malignancy in her parents or two brothers. There is no history of thyroid cancer in any relative. She confirmed with her mother that she had never had childhood therapeutic radiation exposure. She underwent a total thyroidectomy and a 1.8 cm differentiated typical papillary thyroid carcinoma in the left lobe with abundant psammoma bodies, extension into the isthmus, and two positive lymph nodes adjacent to the isthmus were found. No histological findings suggesting residual or an especially aggressive cancer were reported. A 123I scan demonstrated uptake in the right neck, and she received 60 mCi of 131I.
At age 23 years, after ultrasonically suspicious cervical lymph nodes were positive by fine-needle aspiration (FNA) cytology for papillary thyroid cancer with the presence of nuclear inclusions and grooves, she underwent further surgery. Multiple positive lymph nodes and invasion into adipose tissue was found. A 131I scan performed 2 months later demonstrated persistent uptake in the neck and she received 157.6 mCi of 131I after thyroid hormone withdrawal. Afterward, her thyrotropin (TSH) levels were suppressed and thyroglobulin levels remained undetectable. A recombinant human TSH-stimulated 123I whole body scan at age 26 demonstrated no uptake, and magnetic resonance imaging (MRI) of the neck and upper mediastinum at that time was also negative for metastasis. At age 29, she was noted to have suspicious lymph nodes on ultrasound. FNA demonstrated recurrent papillary thyroid cancer, and 3 of 11 lymph nodes removed at surgery were positive. Subsequent ultrasounds did not demonstrate any recurrent disease.
At age 34, a previously unseen lymph node in the left central compartment of the neck was detected by ultrasound. Her thyroglobulin remained undetectable. To evaluate the lymph node further and to look for areas of thyroid cancer, a fluorodeoxyglucose position emission spectroscopy (FDG-PET) computed tomography (CT) examination was performed (Fig. 1). There was no uptake of FDG-18, but a 2.9 cm mass with smooth margins and uniform density, initially suggestive of a cyst, was noted in the upper lobe of the right lung on the CT component of the procedure. The mass did not take up FDG-18 and was not present in the same area visualized on MRI done at age 26. A CT-guided core biopsy of the mass demonstrated a spindle cell neoplasm with features favoring a monophasic synovial sarcoma. A video-assisted thorascopic resection was performed. Since the mass had enlarged from the time of its initial discovery and as the patient had good residual lung capacity, the right middle lobe was resected in addition to the right upper lobe. A 5.4 cm×4.5 cm×2.7 cm mass in the right upper lobe invading into the right middle lobe was removed, with pathology consistent with monophasic synovial sarcoma (Fig. 2). Surgical margins were free of tumor, although the medial margin was only 2 mm as a larger resection was precluded by the presence of vital structures. Immunohistochemical staining was positive for tumor markers CD56 (neural cell adhesion molecule), BCL2 (apoptosis regulator protein), and epithelial membrane antigen (EMA, epithelial surface protein) but negative for CD117 (receptor for activation of tyrosine kinase), S100 (found in malignant peripheral nerve sheath tumors with focal keratin expression), CD34 (endothelial cell marker in solitary fibrous tumors), and TTF1 (a thyroid and lung tissue marker). Total RNA was extracted from the surgical specimen and transcribed into cDNA. Using SSX1 and SSX2 primers with an SS18 primer, the pathognomonic SS18/SSX1 fusion transcript for synovial sarcoma was demonstrated. The patient's post-operative recovery was uneventful. Due to the small surgical margin medially, proton beam therapy was administered, but chemotherapy was not given. There is no evidence of tumor recurrence 10 months after surgery, but she does have persistent dyspnea on exertion. The lymph node noted on previous thyroid ultrasound is no longer present by ultrasound done 12 months after the one that precipitated the discovery of the synovial sarcoma.
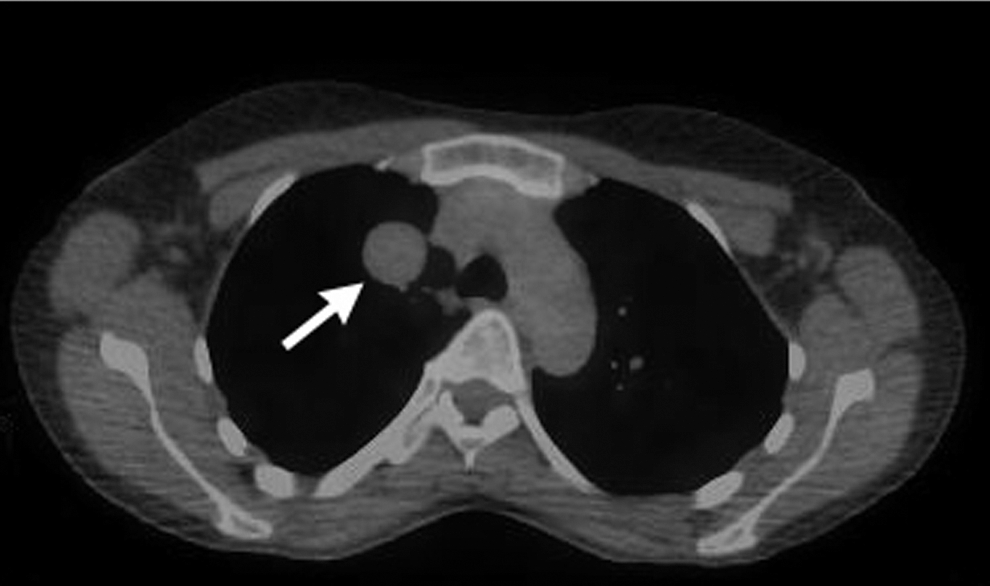
Position emission spectroscopy-computed tomography (PET-CT) examination showing the right upper lobe pulmonary mass (arrow). There was moderate PET enhancement in a left axillary lymph node (standardized uptake value [SUV]=4.08), thought to be due to extravasation of fluorodeoxyglucose seen in the antecubital fossa, but no enhancement of the pulmonary mass.
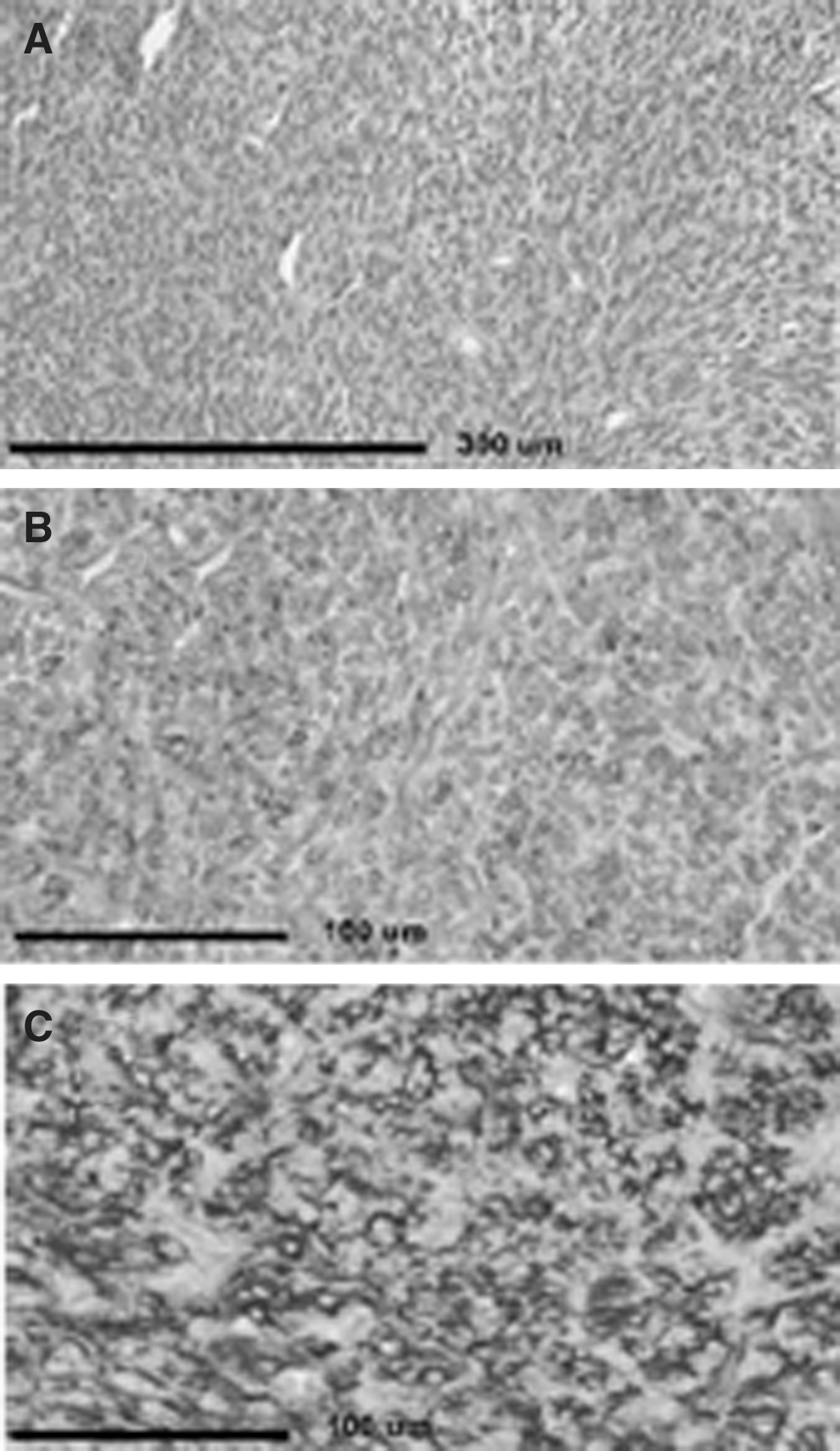
Surgical pathology specimen from right upper lobe lung mass
Discussion
Synovial sarcoma is a soft tissue sarcoma that mainly affects the deep soft tissues of the extremities in adolescents and young adults but can occur in almost any part of the body, including the lungs (5). As reported in 2000, synovial sarcoma represented 5%–10% of all soft tissue sarcomas, with about 800 cases per year in the United States (6). There tends to be a male predominance. There are four histological types: biphasic, monophasic fibrous, monophasic epithelial, and poorly differentiated. The biphasic type consists of epithelial and spindle cell components, whereas the monophasic types have uniform spindle cells and are difficult to distinguish from other spindle cell neoplasms (fibrosarcoma, hemangiopericytoma, leimyosarcoma, spindle cell carcinoma, and carcinosarcoma) (7). In synovial sarcomas, epithelial markers, such as EMA and cytokeratin, are often present (7), but the pathognomonic finding in about 90% of cases is the t(X;18)(p11;q11) translocation (8). This is a rearrangement of the SS18 gene (formerly known as SYT) in the 18q11 region and one of the SSX1, SSX2, or SSX4 genes in Xp11. The rearrangement produces a chimeric SS18/SSX protein in which the last eight amino acids of the SS18 protein are replaced by the carboxy-terminal part of the SSX protein. The functionality of this protein is unclear, but it may have increased transcriptional activity (8). Surgical excision is the primary treatment modality for treatment of synovial sarcoma. Radiotherapy decreases the rate of local recurrence and adjuvant chemotherapy, most commonly a combination of doxorubicin and ifosphamide, given in a few patients has been shown to increase time to local or distal recurrence, but not necessarily improve overall survival (6). In a case review done by Spillane et al. (6), of 150 cases of synovial sarcoma, the 5 year survival rate was 57%. For synovial sarcomas in general, poor prognostic factors included tumor size >5 cm, age >20 years at diagnosis, and the SS18 (SYT)-SSX1 fusion gene. The 5-year progression-free survival rate associated with the SS18-SSX1 gene is 42% versus 89% for the SS18-SSX2 gene (7). However, this may not apply to the very rare pulmonary synovial sarcomas (7).
A review of 60 cases of pulmonary and mediastinal synovial sarcomas demonstrated that the most common presenting symptoms are dyspnea and chest pain (5). Pulmonary and mediastinal synovial sarcomas tend to present in older patients without sex bias. The tumors tend to be more aggressive with 40% presenting as poorly differentiated and an overall 5 year survival rate of 54%. The majority of the patients (58%) had the SS18-SSX1 translocation.
Post-radiation sarcomas have been defined as sarcomas arising in a previously irradiated field after a latency period of at least 2 years (9). Post-radiation sarcomas have been reported with latencies ranging from 3 to 55 years and typically occur after treatment doses ranging from 45 to 60 Gy. Synovial sarcoma after radiation therapy is rare, with only six cases reported in the literature (Table 1). In 1978, Mischler et al. described biphasic synovial sarcoma of the neck after facial and neck radiation for acne, although this case was not confirmed by immunohistochemical or cytogenetic data (10). In 1997, van de Rijn et al. described a 36 year-old woman with synovial sarcoma of the brachial plexus 8 years after receiving chemotherapy and 77 Gy for the treatment of Hodgkin's lymphoma (11). Egger et al. described two cases, the first of which was a 42 year-old woman who developed synovial sarcoma of the minor pectoral muscle 17 years after receiving 171 Gy for breast carcinoma (12). The second patient was a 34 year-old woman who developed synovial sarcoma of the left hand after receiving an unknown dose of radiation to the hand at age 7 for congenital hemihypertrophy secondary to angiomatous malformation. Deraedt et al. reported synovial sarcoma of the left lung in a 20 year-old man with history at age 4 of a metastatic Wilm's tumor that was treated with nephrectomy, pre- and post-operative chemotherapy, and post-operative pulmonary and abdominal radiation (9). Lastly, Mullah-Ali et al. described a paraspinal synovial sarcoma in a 14 year-old girl treated with chemotherapy and 24 Gy to the abdomen and pelvis for neuroblastoma at the age of 14 months (13). The latter five cases of synovial sarcoma were confirmed with analyses demonstrating the t(X;18)(p11.2;q11)) translocation, and all had the monophasic morphology. Four out of six of these cases occurred in women, and the range of latency was 8–27 years.
Age at time of diagnosis of synovial sarcoma.
Grade according to French Federation of Cancer Centers (FNCLCC) grading system (18).
MADDOC protocol: nitrogen mustard, doxorubicin, dacarbazine, cisplatin, vincristine, cyclophosphamide.
CT, chemotherapy, M, male; F, female; mets, metastasis; NA, not available; RT, radiotherapy; SS, synovial sarcoma; RAI, radioactive iodine therapy; LN, lymph node; Tx, treatment; FU, follow-up.
The exact pulmonary exposure from the RAI therapy in the patient described here is not known, as iodine kinetics were not measured, but an estimate can be made. Kolbert et al. evaluated the potential organ doses in thyroid cancer patients treated with RAI therapy (14). They evaluated 26 patients with known metastatic papillary or follicular thyroid cancer and used a combination of PET-CT, 124I scans under TSH alpha stimulation and imaging analysis software to determine the Gray per Gigabequeral (Gy/GBq) for normal organs. Blood testing demonstrated exposure of 0.1 Gy/GBq, and the lung received a mean dose of 0.091 Gy/GBq. Thus, if a patient were to receive 200 mCi of RAI, a normal lung may receive about 0.7 Gy of radiation. Our patient received RAI therapy with thyroid hormone withdrawal (THW); so, the actual delivery of radiation dose to extra-thyroidal tissue was probably >0.7 Gy. Hänscheid et al. randomized 63 patients to RAI therapy with recombinant human thyrotropin (rhTSH) or THW and assessed RAI biokinetics (15). While there was no difference in 48-hours of uptake and residence times in remnant thyroid tissue, rhTSH had significantly longer effective half-life in the remnant tissue and significantly lower absorbed dose to the blood. If we use the ratio observed by Hänscheid et al. (15) for the dose delivered to the blood, we can estimate that the patient's lung received 1.1 Gy.
The patient had not been exposed to ionizing radiation to the pulmonary area other than the RAI therapy. If, in fact, the patient's sarcoma was related to her RAI therapy, why did it occur after a relatively low dose, especially in comparison to other radiation-related sarcomas? It is notable that many papillary thyroid cancers in young people carry RET rearrangements. For example, in 38 cases in children (5–18 years) from Belarus exposed to Chernobyl radiation and 23 sporadic cases in the United States, more than half the children in each group had rearrangements, although the pattern of the translocation sites differed (16). If both of the patient's cancers were the result of genetic rearrangements, perhaps she was particularly susceptible to them. It has been suggested that changes in radiation response genes may increase the risk of thyroid cancer, although the evidence remains inconclusive (17).
Conclusion
We present a patient with history of papillary thyroid cancer treated with RAI therapy who subsequently presented with synovial sarcoma of the lung. Our patient received a total of 217 mCi of 131I, which is within the range reported to cause SPM after RAI (3). While we cannot be sure of the amount of RAI delivery to the lung (pulmonary metastases were never present on imaging), the patient developed a malignancy in the appropriate latency period when compared with other radiation-induced sarcomas. In addition, the development of synovial sarcoma in the lung in proximity to thyroid gland increases suspicion for an association. In summary, considering that the patient was treated for thyroid cancer at a young age and subsequently developed a rare synovial sarcoma with a gene rearrangement, there may have been an association between her RAI treatment and the subsequent development of a synovial sarcoma.
Footnotes
Disclosure Statement
The authors declare that no competing financial interests exist.