
Abstract
Background:
Although thyroid dysgenesis is the most common cause of congenital hypothyroidism (CH), its molecular basis remains largely elusive. Indeed, in only a minority of cases with thyroid dysgenesis (2%–3%) was it possible to identify an underlying genetic defect. The objective of this study was to screen the PAX8 gene and the PAX2 gene in a family with six cases of CH spanning three generations and presenting urogenital malformations. Herein, we report a case series and in vitro characterization of the PAX8 gene mutation.
Methods:
Investigations were conducted at a tertiary care referral center. The index case was diagnosed to have congenital hypothyroidism at 7 months of age when he presented with severe impairment of suckling, constipation, and poor development. Treatment with levothyroxine corrected the symptoms and was associated with catch-up growth. His progeny, including two sons, one daughter, and two granddaughters, were affected by CH, and three of them received the diagnosis at neonatal screening. Ultrasound demonstrated normally located thyroid glands with reduced volumes. Five of the six affected family members, including the index case, had urogenital malformations, including incomplete horseshoe kidney, undescended testicles, hydrocele, and ureterocele. Strabismus was found in three out of six affected patients. No other somatic malformations were found.
Results:
Direct sequencing of the PAX8 gene revealed a new heterozygous mutation (c.74C>G) in all affected individuals. This mutation leads to substitution of proline with arginine at codon 25 (P25R). Fluorescence microscopy showed that P25R is normally located in the nucleus. In transient transfection studies, this mutation causes reduced transcriptional activation ability when using a luciferase reporter construct under the control of a thyroglobulin promoter. This diminished transactivation ability is due to loss of DNA binding capability as shown in electrophoresis mobility shift assay. The sequencing analysis of the PAX2 gene was normal.
Conclusions:
We conclude that this novel PAX8 mutation is responsible for a severe form of dominantly inherited CH. The mutation seems to be associated with abnormalities of the urogenital tract.
Introduction
Within the framework of a statistic survey on the incidence of CH in the Azores Islands (Portugal), a family with six cases of CH across three consecutive generations was identified. The disorder was transmitted in an autosomal dominant manner. All affected individuals had CH. In addition, an unusual occurrence of urogenital malformations was also found in this family. These findings prompted us to screen for PAX8 and PAX2 mutations in this family.
Patients
The pedigree of the family is shown in Fig. 1. The index case (I-1) was born after an uneventful term pregnancy. CH was diagnosed at 7 months of age, when he presented severe impairment of suckling, constipation, and failure to thrive. Once started on levothyroxine, symptoms subsided and a catch-up on growth was noticed. At early puberty, he started complaining of urinary tract infections. Abdominal X rays showed a bilateral renal malformation with an alteration of the renal axis suggesting an incomplete horseshoe kidney. An intravenous urography revealed that both kidneys were located at a lower than the usual position but remained independent one from each other except for the persistence of a fibrous septum between the inferior poles of the kidneys. There was an enlargement of the calyces and bassinets but both ureters were normally implanted although slightly enlarged. A vesicoureteral reflux grade III-IV was evident in tomograms that were obtained later. Except for two episodes of renal stones, the condition remained asymptomatic with abundant water ingestion which forced diuresis. Patient II-1 is the oldest son of the index case. He was born in 1978 after an uneventful term pregnancy (birth weight 3100 g, length 52 cm, Apgar score 9/10). The diagnosis of CH was based on clinical grounds because neonatal screening for CH was introduced later. He was started on levothyroxine 50 μg/d at 5 weeks of life when a confirmatory thyrotropin (TSH) value was above 150 mU/L (normal 0.5–4.5 mU/L). His development was otherwise unremarkable. In patient II-3 who was born in 1981, the diagnosis of CH was also based on clinical grounds because neonatal screening was still not implemented. He was started on levothyroxine 3 weeks after birth. Patient II-5 was diagnosed to have CH through the neonatal screening program, which was introduced in 1982. The initial TSH value obtained from dried blood spot at day 10, was 184 mU/L (normal 0.5–4.5 mU/L), and her thyroxine (T4) level was 3.2 μg/dL (normal 6.5–12.5 μg/dL). She was started on levothyroxine after confirmatory tests. Her daughter, patient III-2 born in 2008, was diagnosed to have CH 6 days after birth through a dried blood spot analysis: TSH, 137 mU/L; T4, 5.6 μg/dL (normal 6.5–12.5 μg/dL). Confirmatory values at day 7 were TSH 187 mU/L; free (F)T4, 0.81 ng/dL (normal 0.89–1.76 ng/dL); thyroglobulin (TG), 47 ng/mL (normal <55 ng/mL). On ultrasound the thyroid gland was located in the normal position but was very small. Urogenital ultrasound revealed an enlargement of the right ureter measuring 8.3 mm at its largest diameter, which was suggestive of a congenital ureterocele. Finally, patient III-1 was the first-degree cousin of patient (III-2) and was born in 2009. CH was diagnosed on the fourth day of life: TSH, 60 mU/L; T4, 2.67 μg/dL (normal, 6.5–12.5 μg/dL); TG, 32 ng/mL (normal, <55 ng/mL) and confirmed on the sixth day of life (TSH, 85 mU/L; FT4, 0.75 ng/mL [normal 0.89–1.76 ng/mL]), when she was started on levothyroxine 50 μg/d. Ultrasound of the thyroid revealed a remnant of a eutopic gland. Her kidneys were normal, but the right ureter measured 9.7 mm in diameter suggesting the diagnosis of a congenital ureterocele. Enlargement of the calyces and bassinets of the homolateral kidney were also noticed. The patient remained asymptomatic, and she continued with normal growth and an adequate intellectual development. Congenital ureterocele was confirmed in both patients (III-1 and III-2) by renal scintiscan 99Tc-diethylene triamine pentaacetic acid.
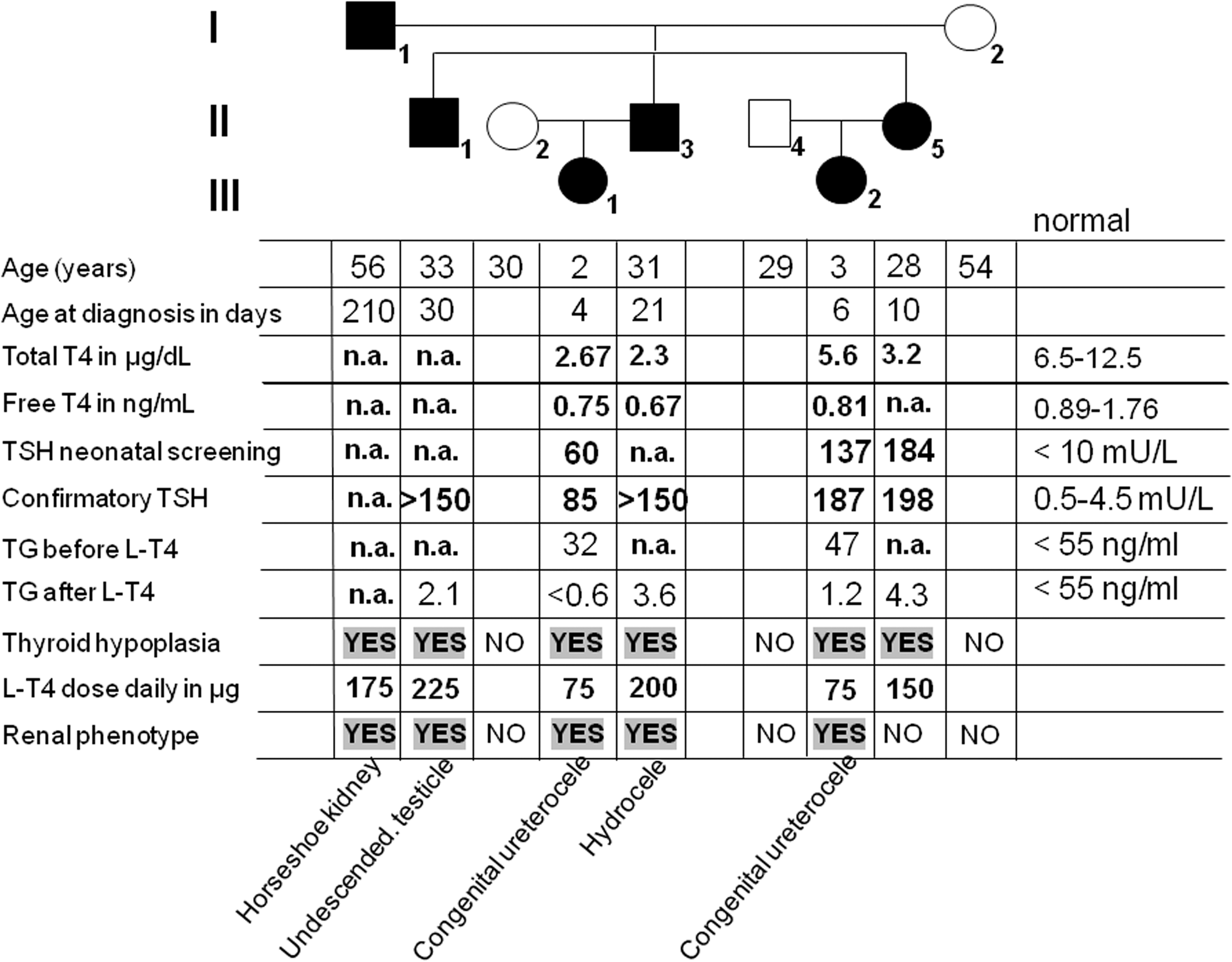
Genealogic tree of the family studied. Each generation corresponds to a roman number. Arabic numbers below each symbol identify the subjects. Clinical and laboratory data are aligned below each symbol. Filled symbols stand for affected individuals who are heterozygous carriers of the mutation. T4, thyroxine; TSH, thyrotropin; TG, thyroglobulin; L-T4, levothyroxine.
Materials and Methods
After obtaining written informed consent, blood was collected and genomic DNA was extracted from peripheral leukocytes utilizing the QIAamp DNA Blood Kit (QIAGEN, Hilden, Germany). The coding sequences of the PAX8 and the PAX2 genes were amplified by polymerase chain reaction (PCR). The primer sequences and PCR conditions are available upon request. The PCR products were purified enzymatically with exonuclease I (New England Biolabs, Frankfurt, Germany) and shrimp alkaline phosphatase (GE Healthcare, Munich, Germany) and then sequenced with BigDye terminator v3 kit using an automated sequencing system (3130 Avant Genetic Analyzer, Applied Biosystems, Darmstadt, Germany).
The construction of the expression vector encoding PAX8 wild-type was described previously (7). The mutations (P25R and F329L) were introduced utilizing the QuickChange Mutagenesis XL kit (Agilent Technologies, La Jolla, CA) according to the manufacturer's protocol. Cloning of PAX8-enhanced green fluorescent protein (EGFP) fusion constructs was performed as described elsewhere (8). The correct sequence of all PAX8 constructs was verified by direct sequencing.
To analyze the activity of wild-type and mutant PAX8, the constructs were transiently transfected into HeLa cells together with a luciferase reporter construct driven by the TG promoter or by the thyroperoxidase (TPO) promoter. Firefly luciferase and renilla luciferase activities were measured 48 hours later with the dual luciferase reporter system (Promega, Madison, WI) according to the manufacturer's recommendations using a Lumat 9507 (Berthold Technologies Bad Wildbad, Germany). All experiments were performed in triplicate and repeated three times. Results are expressed as mean±SEM.
To examine whether the human PAX8-P25R and PAX8-F329L were able to bind one of their target DNA sequences, synthetic oligonucleotides (TPO and TG) were labeled with infrared dye (IRD700, Metabion, Martinsried, Germany) and incubated with in vitro synthesized PAX8 proteins using a reticulocyte lysate kit (Promega) (7). Aliquots of the binding reactions were analyzed by polyacrylamide gel electrophoresis mobility shift assay.
Results
Clinical and laboratorial data are summarized in Fig. 1. Affected members across three generations of the family presented overt hypothyroidism since birth. Three of them were born before implementation of the neonatal screening program. Therefore, the diagnosis of CH was based on clinical grounds and confirmed by blood tests. Failure to recognize the condition during the first weeks of life led to severe impairment of the neurological development as described in the index case. Thyroid ultrasound revealed normally located glands of small size. All but one member of the affected individuals had urogenital malformations, including horseshoe kidney, congenital ureterocele, hydrocele, and cryptorchidism.
Whereas sequencing analysis of the coding sequence of the PAX2 gene revealed no mutation, a heterozygous missense mutation (c.74C>G) that leads to substitution of proline with an arginine at codon 25 (p.P25R) was found in the PAX8 gene. This mutation was not identified in 100 normal alleles of the same genetic background. In addition, another sequence variation (c.987T>C, p.F329L) turned out to be a rare sequence variant since it was found in a heterozygous state in three of the control alleles. In addition, this variant was described as a nonpathogenic allele rs3188996 in the dbSNP database at NCBI. F329L was identified to be located in the same allele as the P25R mutation.
Since PAX8 acts through binding to specific DNA sequences, we analyzed whether the mutant PAX8 protein localizes correctly to the nucleus by fluorescence microscopy. The coding sequences of wild-type PAX8 and the mutant PAX8 P25R and F329L were cloned into EGFP expression constructs and transiently transfected into HeLa cells. All three investigated PAX8 proteins localized correctly in the nucleus (data not shown).
DNA binding was assessed using the PAX8 response elements of the TPO (Fig. 2) and TG (data not shown) promoters with probes labeled with an infrared dye (IRD700) running on a 4% native polyacrylamide gel and visualized with an infrared scanner (Li-Cor Biosciences, Bad Homburg, Germany). The PAX8 wild type and the PAX8-F329L mutation bound normally to DNA, whereas the DNA binding of PAX8-P25R was negligible.

Electromobility shift assay shows that P25R does not bind to the thyroperoxidase (TPO) promoter binding sequence. F329L binds normally to DNA compared to the wild type (WT). As expected, the “double mutant” (P25R-F329L) does not bind to its DNA target sequence. In this experiment the TPO oligo was labeled with an infrared dye (IRD700).
To investigate the functional significance of P25R and F329L, the ability of the mutant protein to activate a cotransfected reporter gene under the control of a human TG (Fig. 3) and TPO (data not shown) promoter was tested. Cotransfection of PAX8 wild type and F329L into HeLa cells produced an increase of luciferase activity with increasing amounts of transfected plasmid DNA, whereas coexpression of the PAX8-P25R mutation produced a negligible activation of the reporter.

Transient transfection studies. PAX8 expression constructs were cotransfected into HeLa cells with a luciferase reporter gene under the control of the thyroglobulin (TG) promoter. The F329L (dark gray bars) construct shows a similar transactivation activity compared to the PAX8 wild type (black bars). In contrast P25R (white bars) and “double mutant” (P25R-F329L; light gray bars) show no transactivational activity.
Discussion
All affected members of the family herein described were heterozygous for a novel PAX8 mutation (p.P25R) which is located in the highly conserved 128–amino acid paired domain. In transient transfection studies, this mutation causes reduced transcriptional activation ability (Fig. 3) due to a loss of DNA binding (Fig. 2). A rare nonpathogenic sequence variant (F329L) was also found in the same allele of the pathogenic P25R mutation. Although normally located, the thyroid glands of the affected patients were small and had low echogenicity in agreement with the overt hypothyroidism presented by the patients since birth. When available, TG levels before initiation of levothyroxine were within the normal range, excluding athyreosis. Hypoplasia of the thyroid glands in these patients is the result of specific properties of PAX8 on both the morphogenesis of the thyroid gland and on the maintenance of the thyroid follicular cells (9). In other patients the gland may be normal at birth but involutes postnatally (10). Finally, patients with PAX8 mutations may present eutopic glands of normal size (11,12). In such cases, other mechanisms of thyroid insufficiency may be more relevant.
Several hypotheses have been raised on how monoallelic PAX8 gene mutations cause such variable phenotypes. One is the possibility of a dominant negative interaction. However, and apart from a few cases (7,13), a dominant negative interaction has not been demonstrated in the majority of 13 different PAX8 mutations already known. Another explanation is related to the effect of gene dosage. The amount of normal PAX8 protein produced by one single normal allele (haploinsufficiency) would not be enough to initiate and maintain the integrity of the normal development (14). The reason is that the PAX8 protein may be one of the contenders in a competition with other transcriptional factors for occupancy of the same DNA response elements. At the level of the TG and TPO promoters, the PAX8 response element overlaps that of TTF1 and binding of the two proteins is somehow mutually exclusive, despite their synergistic effect on TG promoter activation (15). Competition between PAX8 proteins and other transcriptional coactivators and corepressors for common sites in DNA may explain the effect of haploinsufficiency. Alternatively, imprinting of the PAX8 locus may produce a somatic mosaic state responsible by the variability of PAX8 levels not only among individuals but also between tissues (16). It is also possible, that modifier genes play a role in the expression of the phenotype.
The phenotypic particularity of the mutation herein described is the association of CH with urogenital malformations in all but one of the affected individuals. Horseshoe kidney has an incidence of around 1 per 600 live births (17). It is very unlikely that both conditions, horseshoe kidney and thyroid dysgenesis, might have happened in the same patient just by chance. Furthermore, minor alterations of the urogenital tract were found in other members of the family. The PAX8 gene is not only expressed in the thyroid but also in the kidney (6). Together with PAX2, PAX8 is a nephrogenic transcription factor and is required for the formation of the kidneys. However, in only two isolated cases the coexistence of CH and kidney malformations has been reported (18).
In conclusion, we identified and functionally characterized a novel PAX8 gene mutation occurring simultaneously with a rare polymorphism. The mutation leads to thyroid dysgenesis and is associated with urogenital malformations.
Footnotes
Acknowledgments
We thank all members of the family for their willingness to participate in the present study.
Disclosure Statement
The authors have nothing to disclose.