
Abstract
Background:
A randomized phase III trial demonstrated that vandetanib treatment is effective in patients with metastatic medullary thyroid cancer (MTC), leading to regulatory approval, but its use may be associated with toxicities that require specific monitoring and management. The objective of the present study performed in France was to describe the toxicity profile and efficacy of vandetanib treatment when given outside any trial.
Methods:
Sixty-eight patients were treated with vandetanib in the frame of a temporary use authorization (ATU) in France from August 2010 to February 2012, when the drug was available on request for patients with locally advanced or metastatic MTC. Patients were registered by the French health authorities, and characteristics, treatment parameters, toxicity profile, and efficacy were retrospectively reviewed. Eight patients were excluded from the analysis because vandetanib treatment was not administered (n=3), had been given in a trial before ATU (n=3), or was given for a non-MTC cancer (n=2).
Results:
Data from the 60 MTC patients were analyzed. Mean age was 58 years (range 11–83 years), 39 patients were male, and six had hereditary MTC. Fifty-six (93%) had metastatic disease in the mediastinum (82%), bones (65%), liver (53%), or lung (53%), and four had only locally advanced disease. At the time of study evaluation, with a median follow-up of 20 months and a median duration of treatment of 9.7 months (range 0.3–36 months), 15 patients were continuing vandetanib treatment (range 18–36 months). Median progression-free survival was 16.1 months. Twenty-five patients discontinued treatment for disease progression (range 0.3–29 months). Best tumor response was a complete response in one patient, a partial response in 12 (20%), stable disease in 33 (55%), and progression in seven patients (12%). All patients had at least one adverse event (AE) during treatment. The main AEs were skin toxicity, diarrhea, and asthenia. Sixteen patients (27%) discontinued treatment for toxicity, and one patient died from vandetanib-induced cardiac toxicity.
Conclusions:
Vandetanib is an effective option for patients with advanced MTC. AEs should be monitored carefully and should be minimized by educating both patients and care providers and by applying symptomatic treatment and dose reduction.
Introduction
V
In Europe, vandetanib is indicated for aggressive and symptomatic MTC, a subgroup of patients recruited to the ZETA phase III registration trial. The ZETA trial reported notable toxicities, including hypertension, diarrhea, fatigue, skin toxicities (rash, folliculitis, photosensitivity), and prolongation of the QT interval on electrocardiogram that may lead to “torsades de pointes” and sudden death. Commitments associated with the approval of the drug included prevention of toxicities by education of care providers and patients and their management with appropriate symptomatic treatment modalities and dose reduction/discontinuation.
To the authors' knowledge, approximately 2160 patients with MTC have been treated with vandetanib, of whom 565 were treated in clinical trials. To date, no data have been published about the use of vandetanib in routine clinical practice. The objective of the present study performed in France was to describe the toxicity profile and efficacy of vandetanib treatment when given outside any trial. In France, there was an opportunity to collect safety data on the use of vandetanib in real-life practice in all MTC patients who were given the drug under the umbrella of a temporary use authorization (ATU). The ATU is active for a limited period of time during which a treatment may be given for a specific disease for which there is no alternative treatment modality outside of a therapeutic trial, and it covers the time between EMA approval and acceptance of the drug by French authorities. During this time period, all patients treated with the drug under the umbrella of the ATU are registered. Patients are selected for treatment according to the indications of the EMA label, and are then treated and followed according to local practices based on the recommendations of a multidisciplinary board. The present study is a retrospective analysis of all patients who were enrolled in the vandetanib ATU in France. The objective of this study was to describe the toxicity profile of and response rate to vandetanib treatment in real-life practice in France and outside the frame of a controlled trial.
Patients and Methods
Inclusion criteria
Sixty-eight patients were treated with vandetanib from August 2010 to February 2012 under the umbrella of the ATU with all the files available for review. Selection criteria for treatment were based on international guidelines (2,3) and were in agreement with the ongoing indications specified in the EMA label: MTC patients with locally advanced disease not amenable to local treatment modalities or with distant metastases, who had a large tumor burden, and either with documented progression within 12 months or with symptoms. For most patients, the decision to initiate treatment was validated by a multidisciplinary thyroid tumor board of the French Tumeurs de la THYroïde REFractaires (TUTHYREF) network. During this time period, each patient who was going to be treated with vandetanib was recorded. The treating physician had to provide written information on clinical, biological, and morphological characteristics for registering the patient, and then updated the information every two months to continue the treatment. Patients were treated in each center according to local practice, and toxicity was recorded and was managed locally by the treating physician. Efficacy was assessed locally by serum calcitonin and carcino-embryonic antigen (CEA) determinations, and by imaging that included computed tomography scans every two to three months (local assessment, not necessary RECIST criteria). Additionally, magnetic resonance imaging of the liver and bone scintigraphy were performed in some patients.
An independent retrospective review of the ATU data was conducted between May and December 2013, by reviewing the clinical file of each patient. This comprised the clinical characteristics of the patients and treatment parameters, including its toxicity profile and its efficacy.
Exclusion criteria
Eight patients were excluded from the analysis: two patients who were treated for a cancer type that was not a MTC (one follicular thyroid cancer and one malignant teratoma), three patients who had already been treated with vandetanib in the phase III ZETA trial, and three patients who did not receive vandetanib (two patients died before initiation of treatment and one patient finally refused to be treated with vandetanib). The remaining 60 patients constituted the basis of the present analysis.
Statistical analysis
Descriptive analyses were performed using means and standard deviations for quantitative variables. Qualitative variables are expressed as percentages. Progression-free survival (PFS) is defined as the delay between the date of initiation of vandetanib treatment and the occurrence of an event defined as either progression or death. Patients who did not have an event were censored at the date of last follow-up. PFS rates were estimated by the Kaplan–Meier method. The software used was SAS version 9.1 (SAS Institute, Inc., Cary, NC).
Results
Patients
The 60 MTC patients (39 males) who were treated with vandetanib under the umbrella of the ATU were analyzed (Table 1). The median age of patients at diagnosis of MTC was 48 years (range 8–82 years), and the median age at initiation of vandetanib treatment was 58 years (range 11–83 years). The majority of patients (48; 80%) presented with sporadic disease, and six patients had an hereditary form of the disease (multiple endocrine neoplasia type 2A and type 2B). The genetic status was unknown for six patients.
MEN, multiple endocrine neoplasia; MTC, medullary thyroid carcinoma.
At the initiation of vandetanib treatment, symptoms were present in 13 (22%) patients and included diarrhea in 10. ECOG status was 0 or 1 in 51 patients, and 2 or 3 in seven patients. Previous treatment modalities included neck surgery in 49 patients and neck external beam radiation therapy in 22 patients. One patient was treated only with neck external irradiation. Eleven patients (18%) had received another systemic treatment before initiation of vandetanib treatment: six had been treated with another tyrosine kinase inhibitor in a clinical trial (cabozantinib in five patients, lenvatinib in one patient), and five patients had been treated with cytotoxic chemotherapy (two with 5-fluorouracil (FU)-dacarbazine, one with 5 FU alone, one with cisplatin-etoposide, and one with the FOLFOX regimen: 5-FU+oxaliplatin). Vandetanib was the first systemic treatment for 49 patients (82%).
At vandetanib initiation, four patients (7%) had unresectable, locally advanced disease with no known distant metastases, and 56 patients (93%) had distant metastases. Forty-four patients (74%) had local disease, consisting of the primary tumor in four patients (7%) and a neck recurrence in 40 patients (67%). Distant metastases were located in the liver in 32 cases (53%), in the lung in 32 cases (53%), in the mediastinum in 47 cases (78%), in the bone in 39 cases (65%), and in other sites (kidney, skin, brain, pericardium, choiroidum, and axillar lymph nodes) in 14 cases (23%). The metastatic spread involved none, one, two, or three or more sites in four (7%), 7 (12%), 20 (33%), and 29 (48%) cases, respectively.
Radiological evaluation within the year before treatment initiation was available in 51 cases and showed disease progression in 44 patients (73%). In fact, the morphological progression before vandetanib treatment could not be assessed precisely in all patients because of differences in the imaging procedure and variability in the frequency of morphological assessments.
The initial vandetanib treatment dose was 300 mg/day in 55 patients, and was lower in the five patients with ECOG grades 2 or 3. The median follow-up was 20 months, ranging from 0.3 to 36 months from the initiation of vandetanib treatment. At the end of the data collection, 15 of the 60 patients were still receiving treatment.
Efficacy
The median duration of vandetanib treatment was 9.7 months (range 0.3–36 months). Vandetanib treatment was given for more than six months to 38 patients (63%), for more than 12 months to 25 patients (42%), and for more than 24 months to 15 patients (Fig. 1). Thirty-eight patients had no vandetanib dose reduction. Vandetanib dosage was decreased in 20 patients (33%), and dose reduction was unknown for two patients. The vandetanib dose at the end of data collection was 300 mg/day for 38 patients, 200–250 mg/day for 12 patients, 100–150 mg/day for seven patients, and 50 mg daily for one patient. Among the 15 patients still on treatment at the end of data collection, 10 were still treated with 300 mg/day, whereas five patients had had their dose reduced, two to 100 mg daily for and three to 200 mg daily. As vandetanib is supplied in a 300 mg tablet or a 100 mg tablet, the other doses were given by administering 100 mg every other day to provide 50 mg daily on average, 300 mg every other day to provide 150 mg daily on average (or alternating 100 mg with 200 mg per day), or alternating 200 mg with 300 mg per day to provide a 250 mg daily average.
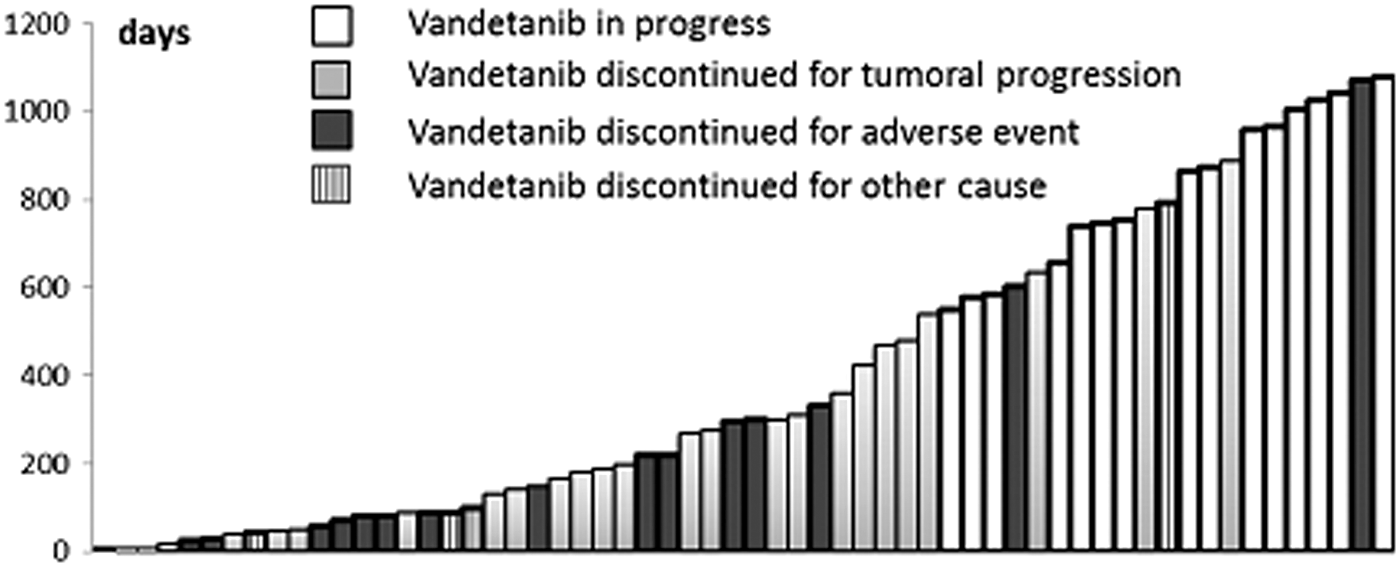
Duration of treatment with vandetanib (in days) and cases of discontinuation. Note: One bar represents each patient.
Morphological evaluation was available for 53 patients; five patients died from disease progression before any morphological evaluation and two were lost to follow up. Among the 60 patients (intention to treat), the best morphological response was a complete response (CR) in one patient (2%), a partial response (PR) in 12 (20%), stable disease (SD) in 33 (55%) that was longer than 23 weeks in 23 patients (38%), and progressive disease (PD) in seven patients (12%). Thirteen patients experienced clinical improvement of their symptoms (decrease of diarrhea or local cervical discomfort) after one month of treatment.
Biological evaluation was available for 50 patients. Serum calcitonin decreased by 50% or more in 33/50 cases (70%) in whom calcitonin was measured, and CEA level decreased by 50% or more in 19 of the 41 patients (46%) with an initially abnormal CEA level.
The median PFS was 16.1 months (Fig. 2). At the end of data collection, 25 patients had died (median time to death: 12 months after treatment initiation; range 0.3–26 months), all deaths except two being related to disease progression. The one-year survival rate for the 60 patients was 78% (47/60), and the two-year survival rate was 60% (36/60).

Progression-free survival from the time of vandetanib initiation. Note: 15 patients were still being treated at the end of data collection.
Safety and tolerability
Vandetanib was discontinued because of disease progression in 25 patients (42%), including two patients who died 10 days after the initiation of vandetanib treatment, with a median duration of vandetanib treatment of 9.7 months (range 0.5–30 months). Vandetanib was discontinued because of adverse events (AEs) in 16 patients (27%), with a median duration of vandetanib treatment of four months (range 0.3–36 months). Vandetanib was discontinued for other reasons in four cases: one patient with chronic myeloid leukemia and another patient with myocardial infarction (both events were considered unrelated to vandetanib), and two patients decided to discontinue vandetanib treatment with a median duration of vandetanib treatment of one month (Fig. 1).
The most common AEs (any grade) are summarized in Table 2. Skin toxicity occurred in 45 patients (75%) and included folliculitis/rash/dry skin in 36 (60%), photosensitivity in 13 (22%), and hand/foot skin reaction in seven (12%). Gastrointestinal toxicity occurred in 37 cases (62%), including diarrhea in 35 (58%) and digestive occlusion in two (3%). Asthenia occurred in 31 cases (52%). Prolongation of the QT interval on electrocardiogram (ECG) occurred in 19 patients (32%): two patients with grade 2 (QTc prolongation >480 msec) and three patients with grade 3 (QTc prolongation >501 msec), including one patient who developed an arrhythmia (grade 4). Vision abnormalities occurred in six patients (10%), including fuzzy vision, phosphene, blue halo, and corneal deposits. These last two events occurred in two cases at 12 and 15 months after treatment initiation.
Adverse events were graded according to National Cancer Institute Common Terminology Criteria of Adverse Events, version 4, after reading the patient's file. Percentages are expressed for intention to treat (60 patients).
Sixteen patients (27%) discontinued vandetanib because of a serious AE. Gastrointestinal toxicity leading to treatment discontinuation included diarrhea in six cases, diverticulitis in one case, and hypokalemia and QT prolongation induced by grade 4 diarrhea in one patient. This latter patient developed an arrhythmia that was responsible for the patient's death. Other events included hemoptysis after fistula formation in one case, neurological toxicity leading to treatment discontinuation in three cases, meningeal hemorrhage in one patient, a transient ischemic accident in one patient, and transient Wernicke aphasia in one patient. Skin toxicity led to treatment discontinuation in two patients and included grade 3 folliculitis in one case and Lyell syndrome in a second patient. Finally, asthenia led to treatment discontinuation in four cases.
Discussion
This is the first report on the use of vandetanib in routine clinical practice outside the context of a controlled clinical trial. No specific protocol was used, and patients were treated according to local clinical practice. Almost all French patients with locally advanced or metastatic MTC with large tumor burden who had either symptomatic and/or progressive disease were enrolled in the ATU because at that time there was no other treatment available for these patients. During an 18-month period, only 60 MTC patients were treated with vandetanib, indicating that in France, with a global population of 66 million individuals, the annual number of MTC patients who could be treated with vandetanib (or any other available drug) is about one patient per one and a half to two million individuals. Of the 60 MTC patients, 51 (85%) were treated at a center belonging to the TUTHYREF network, the French network for refractory thyroid cancers. Therefore, the decision to treat was validated for most patients by a national tumor board.
Patients were registered prospectively, and for the purpose of the present study, all clinical data from all patients were then retrospectively reviewed. This study confirms the efficacy of vandetanib reported in previous phase 2 and phase 3 studies, in terms of both PFS and objective response rate (1,4). Differences with previous reports in terms of PFS and RR may be not significant or related in part to the selection of patients or the assessment of efficacy. Most patients had advanced and progressive or symptomatic disease. They had similar characteristics to those included in controlled trials with vandetanib in terms of age and spread of disease, but had most likely a more severe clinical picture. Patients were in close agreement with the EMA label and more similar to those included in controlled trials with cabozantinib (5), but with more aggressive behavior. The patient with complete response presented with an initial pT4bN1b (intratracheal) MTC, and vandetanib was given because of several large upper mediastinal lymph nodes (one retro-tracheal of 74 mm and three latero-tracheal lesions; not biopsy proven) with dyspnea, and a baseline calcitonin of 61 pg/mL. Complete morphological response was demonstrated after five months of treatment and persisted at two and a half years after initiation of vandetanib treatment.
This study also confirms that adverse events are frequent and that most are manageable with dose reduction and symptomatic treatments. Dose reduction was necessary in 33% of patients and drug discontinuation in 27% of patients, in agreement with previous studies using other TKIs and twice as frequent as in previous studies using vandetanib (1), probably due to medical inexperience. In the vast majority of cases, vandetanib was used for the first time by the treating physician (only few had previously used vandetanib in the context of trials). The incidence of asthenia and QT prolongation was higher than in the ZETA trial. The likely causes are that routine ECG/electrolyte/thyrotropin monitoring and control was not as tight as in a trial and it was in part not stated whether the adverse events were related to the drug therapy. One patient died from cardiac arrhythmia after eight months of vandetanib treatment. This was related to severe hypokalemia (2 mmol/L measured in the blood sample obtained with difficulty in the intensive care unit, but with an U wave on ECG and a QTc of 670 msec in the emergency unit) that was induced by severe diarrhea that persisted over several weeks and that could not be controlled.
Median treatment duration was 42 weeks. This is also in agreement with previous data, demonstrating that once this treatment has been initiated, it can be given for prolonged periods of time. The fact that patients may undergo long-term treatment shows that only selected MTC patients with advanced disease should be treated with vandetanib, that is, those with large tumor burden and with either documented progression within a year, or patients presenting with tumor-associated symptoms. The decision to initiate treatment with a TKI such as vandetanib is optimally validated by a multidisciplinary team. Because of the long duration of treatment, side effects should be minimized and quality of life should be maintained. Also, serious adverse events should be avoided, and in the majority of patients, prolongation of the QT interval may be prevented by maintaining serum electrolytes (potassium, calcium, and magnesium) within their reference ranges, and by avoiding administration of other drugs that may further prolong the QT interval (6).
In conclusion, this retrospective study on the use of vandetanib in routine clinical practice and outside any clinical trial confirms data obtained in the frame of a controlled trial in terms of efficacy. Even more importantly, it does not demonstrate any unexpected toxicity. Indeed, notable toxicities exist, but they are manageable by optimizing education of care providers and patients.
Footnotes
Author Disclosure Statement
CC: advisory board, after the completion of the study, with AstraZeneca. TUTHYREF network received research support from AstraZeneca. MS: consulted and received research support from AstraZeneca, and honoraria.