
Abstract
Background:
Monocarboxylate transporter 8 (MCT8) is a thyroid hormone-specific cell membrane transporter. Mutations in the MCT8 gene lead to profound psychomotor retardation and abnormal thyroid hormone serum levels with low thyroxine (T4) and high triiodothyronine (T3). Currently, therapeutic options for patients are limited. Triiodothyroacetic acid (TRIAC) has potential therapeutic value. The aim of this study was to evaluate the effects and efficacy of therapeutic doses of TRIAC on Mct8-deficient mice (Mct8KO).
Methods:
Wild-type (Wt) and Mct8KO mice were treated with 30 ng TRIAC/g of body weight/day, given in drinking water, from postnatal day 21 to 30. TRIAC, T4 and T3 levels in plasma, as well as T3 and TRIAC content in the cerebral cortex and striatum were measured by specific radioimmunoassays. The activities of deiodinases 1 and 2 were measured in liver and cortex. The effect of TRIAC treatment in the expression of T3-dependent genes was measured in the heart, cerebral cortex, and striatum.
Results:
Plasma TRIAC concentration were the same in Wt and Mct8KO animals after treatment. TRIAC treatment greatly decreased plasma T4 in Wt and Mct8KO mice, and reduced T3 to normal levels in the Mct8KO mice. Deiodinase 1 activity and gene expression in the liver increased, while it did not have any effect on the expression of Serca2a in the heart. TRIAC treatment did not induce the expression of T3-dependent genes in the cerebral cortex or striatum, but further decreased expression of Flywch2 in the cortex and Aldh1a1 and Flywch2 in the striatum. Direct measurements of TRIAC and T3 content in the cortex and striatum revealed a decrease in T3 after treatment with no significant increase in the level of endogenous TRIAC.
Conclusions:
Therapeutic doses of TRIAC in Mct8KO mice restored plasma T3 levels but severely decreased T4 levels. TRIAC has a direct effect on deiodinase 1 in the liver and does not have an effect on gene expression in the heart. The increase in the plasma TRIAC levels after treatment is not sufficient to increase TRIAC levels in the brain and to promote the expression of T3-dependent genes in brain cells. Instead, it leads to a state of brain hypothyroidism with reduced T3 content.
Introduction
T
Mct8 knockout (Mct8KO) mice were generated as a model for Allan–Herndon–Dudley syndrome. These mice faithfully replicate the alterations in the TH concentrations in serum of the patients (18,19). However, they do not present gross neurological abnormalities (20). Regardless of this, they have been a useful tool to understand some aspects of the syndrome and to explore the differences between mice and humans. Studies using these mice have demonstrated that despite having high serum T3 concentrations, Mct8KO mice have a reduced T3 content in the brain (18,19). Also, it has been suggested that the pathogenesis associated with MCT8 deficiency arises from an impaired T3 transport through the blood–brain barrier (21). Recent studies of brain sections have provided evidence for cerebral hypothyroidism in MCT8-deficient patients (22). Therefore, MCT8-deficient patients would paradoxically present peripheral hyperthyroidism in peripheral tissues and brain hypothyroidism with associated neurological impairments.
The therapeutic options for these patients are rather limited. Patients have been treated with levothyroxine (LT4), but not only did this approach not improve neurological function, but it also further increased the serum T3 levels (10,23). To avoid the latter, propylthiouracil (PTU) has been administered together with LT4. This treatment allows T3 levels to be normalized in peripheral tissues, improving the thyrotoxic state, but it did not improve the neurological condition (24,25). In another approach, four young MCT8-deficient patients were treated with the TH analogue 3,5-diiodothyropropionic acid (DITPA). Although this treatment normalized the serum TH levels, ameliorating the thyrotoxicosis of peripheral tissues, no improvements in psychomotor development were observed (26).
The natural TH metabolite 3,3′,5-triiodothyroacetic acid (TRIAC) presents some characteristics that make it a good candidate to use it as a therapeutic agent in MCT8 deficiency. For instance, TRIAC binds with the same affinity as T3 to the TH receptor alpha and with higher affinity to the TH receptor beta (27). Also, there is extensive clinical experience with TRIAC, as it has successfully been used in patients with thyroid cancer (28) and with resistance to TH (29,30). Most importantly, in vitro and in vivo studies in mice have demonstrated that (i) TRIAC is transported into brain cells by other transporters than MCT8; (ii) it is generated from Tetrac by type 2 deiodinase (D2) activity; (iii) it is metabolized by type 3 deiodinase (D3) in the same way as T3; (iv) it induces similar neuronal gene responses as T3; and (v) TRIAC is able to restore neuronal differentiation in the hypothyroid brain (31,32). However, the fact that TRIAC produces similar neuronal gene responses as T3 was assessed only in vitro, and the in vivo studies concluding that TRIAC restores neuronal differentiation in the hypothyroid brain in the athyroid Pax8 knockout mice, and the double knockout for Mct8 and the organic anion transporter polypeptide 1 C1 (Oatp1c1) were obtained after treatment with high doses of TRIAC (200–400 ng/g of body weight [BW]), while lower doses of 50 ng/g BW had modest effects (32).
Previous studies in rodents have demonstrated that TRIAC treatment induces a decrease in T4 and T3 levels in plasma (33). It is unclear what the effects of therapeutic doses of TRIAC might be in MCT8 deficiency. Moreover, it is unknown if therapeutic doses of TRIAC are able to induce similar neuronal gene responses as T3 in the brain in vivo. Mct8KO mice provide a good model to study these aspects. To begin with, as Mct8KO replicate the patient's endocrine syndrome, it is possible to assess the effect of TRIAC on serum TH levels. Also, although Mct8KO mice do not faithfully replicate the severe neurological impairments, they present some alterations in the expression of T3-dependent genes that can be used as markers to evaluate TRIAC action in the brain (34). Finally, Mct8KO mice have already been proven to be a good model to study the effect of other TH analogues as possible treatments for MCT8 deficiency (35,36).
In human clinical trials, TRIAC has been tested at doses ranging from 5 to 50 μg/kg BW/day (28 –30). This study evaluated the effect of therapeutic doses of TRIAC (30 ng/g BW/day) on Mct8KO mice to characterize the effect of TRIAC as a potential therapeutic agent in MCT8-deficient patients. A state of hypothyroidism was not induced. Hence, the basal conditions are more similar to those in MCT8-deficient patients. Although a prenatal diagnosis of MCT8 is possible and has already been reported for male fetuses in pregnant women heterozygous for MCT8 mutations (37), most patients are diagnosed after birth. For this reason, the study was performed in juvenile mice in order to have comparable conditions. Because patients suffer from peripheral tissue hyperthyroidism, the aim was to assess the effects of TRIAC on plasma TH levels and T3-dependent gene expression in the liver and heart. Previous treatment with other compounds has been successful in normalizing serum TH levels, improving the signs and symptoms that arise from peripheral tissue thyrotoxicosis, but they have failed to improve the neurological disorder. The ultimate goal of this work was to evaluate TRIAC activity in the CNS. This was done by evaluating its effects on T3-dependent gene expression and by measuring TRIAC and T3 content in the cerebral cortex and the striatum.
Materials and Methods
Ethics statement
All experimental procedures involving animals were performed following the European Union Council guidelines (directive 2010/63/UE) and Spanish regulations (R.D.1201/2005, and Law 32/2007), and were approved by the ethics committee at Consejo Superior de Investigaciones Científicas (CSIC; approval number ES280790000188). All efforts were made to minimize suffering, as indicated below.
Experimental animals
Animals were housed in temperature- and light-controlled conditions at 22 ± 2°C on a 12:12 light–dark cycle (lights on at 8am), and they had access to food and water ad libitum. Mct8 (Slc16a2) KO mice (male genotype, Mct8–/y ) were originally produced by Dumitrescu et al. (18) by homologous recombination. Experiments were carried out with Mct8+/y and Mct8–/y mice from the same litters derived by backcrossing heterozygous females with Wt males of the C57BL/6J strain at the authors' animal facility. The pups were genotyped on postnatal day (P) 18 to select for Mct8+/y and Mct8–/y mice from the same litters. For simplicity, these animals will be referred to as Wt and Mct8KO mice, respectively. The Mct8 genotype was confirmed as described (21).
At P21, concurring with the weaning, the pups were given either drinking water or TRIAC (Sigma-Aldrich) in the drinking water until P30. The TRIAC concentration was calculated on the basis of fluid intake to provide 30 ng/g BW/day in the drinking water containing 0.01% bovine serum albumin. Animals were killed at P30, two hours after the end of the dark cycle, when most of the water intake takes place.
In one set of experiments, the following groups were prepared: Wt mice (n = 5), Mct8KO mice (n = 7), Wt mice treated with TRIAC (n = 5), and Mct8KO mice treated with TRIAC (n = 7). The animals were killed by decapitation. Trunk blood was collected in heparinized tubes after decapitation and was used for T3, T4, and TRIAC determinations in plasma. The brain was removed, and the cerebral cortex and the striatum were rapidly dissected out from the underlying structures, blotted on filter paper, frozen on dry ice, and stored at −80°C. The liver and heart were also harvested. These tissues were used for gene expression analysis.
In another set of experiments, Wt mice (n = 7), Mct8KO mice (n = 10), Wt mice treated with TRIAC (n = 8), and Mct8KO mice treated with TRIAC (n = 11) were anesthetized with ketamine (50 μg/g BW) and medetomidine hydrochloride (0.1 μg/g BW), and perfused with saline to remove blood from tissues before their collection. Prior to perfusion, blood was extracted by retroorbital collection and used for the determination of TRIAC and T4 and T3 plasma concentrations in order to verify TRIAC's effects on plasma TH concentrations. The brain was removed, and the cerebral cortex and the striatum were rapidly dissected and used to determine the tissue T3 and TRIAC content.
Gene expression
RNA was isolated from individual hemicortices, striatum, liver, and heart. The expression of the following T3-dependent genes was measured by quantitative polymerase chain reaction (qPCR) in the cerebral cortex and the striatum: Hr (Hairless), Cbr2 (carbonyl reductase), Itih3 (inter-trypsin inhibitor, heavy chain 3), Aldh1a1 (aldehyde dehydrogenase 1 family, member A1), and Flywch2 (FLYWCH family member 2); in the liver: Dio1 (type 1 iodothyronine deiodinase); and in the heart: Serca2a (sarcoplasmic reticulum Ca2+ ATPase pump). Procedures for RNA extraction and qPCR were identical to those previously described (34). Data were expressed relative to the values obtained on tissues from the Wt mice in basal conditions, which were given a mean value of 1.0 after correction for 18S RNA.
Radioimmunoassays of T4, T3, and TRIAC in plasma and tissues
TRIAC, T3, and T4 concentrations in plasma, as well as TRIAC and T3 content in tissues, were determined as previously described (33,38). Individual 120-μL aliquots of plasma were used, and both hemicortices and striatum of individual mice and the samples were extracted for radioimmunoassay (RIA) determination. T3 data were corrected for the cross-reactivity of TRIAC on the T3 antiserum (17%). The TRIAC antiserum was a kind gift from Dr. Albert Burger.
D1 and D2 enzymatic activities
D1 activity in liver and D2 activity in the cerebral cortex were measured as described (33). Data were analyzed by two-way analysis of variance and Bonferroni's post hoc test using GraphPad software (
Results
Effects of TRIAC in plasma
The administration of 30 ng of TRIAC/g BW/day from P21 to P30 caused nearly a threefold increase in plasma TRIAC levels compared with their basal controls, and reached the same concentration in both the Wt- and Mct8KO-treated mice. This shows that administration of the same dose of TRIAC to both genotypes results in the same plasma concentration of this analogue (Fig. 1).

Triiodothyroacetic acid (TRIAC), thyroxine (T4), and triiodothyronine (T3) plasma levels (M ± standard error [SE]) in wild type (Wt) and Mct8 knockout (Mct8KO) mice in basal conditions and after TRIAC treatment. **p < 0.01, and ***p < 0.001 were determined by two-way analysis of variance (ANOVA) and Bonferroni's post hoc test, the two factors being genotype and treatment.
As previously reported (18,19), plasma T4 levels were decreased in the basal Mct8KO animals to nearly half the levels of the basal levels of the Wt. TRIAC treatment drastically reduced the plasma T4 levels in both the Wt and the Mct8KO. T4 levels were reduced by 3.5-fold in the treated animals compared with their respective basal controls, and this decrease resulted in a sixfold difference between basal Wt- and TRIAC-treated Mct8KO animals. TRIAC is a well-known inhibitor of TSH (39 –41), consequently decreasing plasma T4 levels (33). Although the TSH levels were not measured, the data suggest that Mct8 deficiency does not interfere with the feedback regulation of TSH by TRIAC causing a decrease in the plasma T4 levels, as in the Wt animals (Fig. 1).
As expected, basal plasma T3 levels were significantly higher in Mct8KO compared with Wt animals. Most importantly, TRIAC treatment decreased T3 to normal levels in the Mct8KO and did not affect T3 levels in the Wt mice (Fig. 1).
Effects of TRIAC on the liver and heart
The study of D1 enzymatic activity in the liver revealed the already described increase of this enzyme in Mct8KO animals compared with the Wt animals (18,19). TRIAC treatment increased D1 activity so that levels in the Wt and Mct8KO animals were no longer different (Fig. 2). This occurred despite a decrease in the plasma T3 level and was due to the thyromimetic effect of TRIAC.

Liver D1 enzymatic activity and gene expression of Dio1 in the liver and Serca2a in the heart (M ± SE) in Wt and Mct8KO mice in basal conditions and after TRIAC treatment. Gene expression measurements were obtained by quantitative polymerase chain reaction (qPCR), and the data are expressed relative to 18S RNA. **p < 0.01, and ***p < 0.001 were determined by two-way ANOVA and Bonferroni's post hoc test, the two factors being genotype and treatment. Dio1, type 1 iodothyronine deiodinase mRNA; Serca2a, sarcoplasmic reticulum Ca2+ ATPase pump mRNA.
Consistent with the D1 activity results, Dio1 mRNA expression was increased in the Mct8KO compared with the Wt animals. TRIAC treatment increased the Dio1 expression fivefold in the Wt mice and twofold in the Mct8KO mice compared with their basal levels (Fig. 2), confirming the thyromimetic activity in the liver.
Taken together, the findings indicate that TRIAC, similar to T3, has a direct effect on the liver, regulating Dio1 expression and D1 activity.
With respect to the heart, TRIAC treatment did not have an effect on expression of Serca2a under any of the experimental conditions (Fig. 2).
Effects of TRIAC in the CNS
As MCT8-deficient patients present severe CNS deficits consistent with TH deficiency, the effect of therapeutic doses of TRIAC on the CNS of Mct8KO animals was further analyzed to evaluate its potential use as a therapeutic agent.
To explore if the therapeutic doses of TRIAC used in this study reach the brain and are able to exert any function at the genomic level, the expression of T3-dependent genes were studied in two different regions of the brain: the cerebral cortex and the striatum. The selected genes were previously identified to be altered in Mct8 deficiency, such as Hr and Cbr2 (34), which are known T3-responsive genes (42). The expression of some genes in mice cerebral cortex and striatum that are not altered in Mct8-deficient mice, but that are also T3-responsive genes, such as Itih3, Aldh1a1, and Flywch2, were also examined (42).
In the cerebral cortex (Fig. 3, left panels), the differences between Wt and Mct8KO animals at basal conditions were consistent with previous studies: the expression levels of Hr and Cbr2 were decreased in the Mct8KO animals compared with the Wt controls, while the expression levels of Itih3, Aldh1a1, and Flywch2 did not vary between the Wt and the Mct8KO mice (34). TRIAC treatment did not increase the expression of Hr or Cbr2 in the Wt or the Mct8KO animals after TRIAC treatment, suggesting that the treatment is not able to correct the gene expression defect of the Mct8KO mice. Moreover, TRIAC treatment did not increase the expression levels of Itih3 and Aldh1a1 in either the Wt or the Mct8KO animals compared with their basal controls. Flywch2 expression revealed no changes after treatment in the Wt animals, but contrary to expectations, there was a decrease in its expression levels in the TRIAC-treated Mct8KO animals compared with baseline.
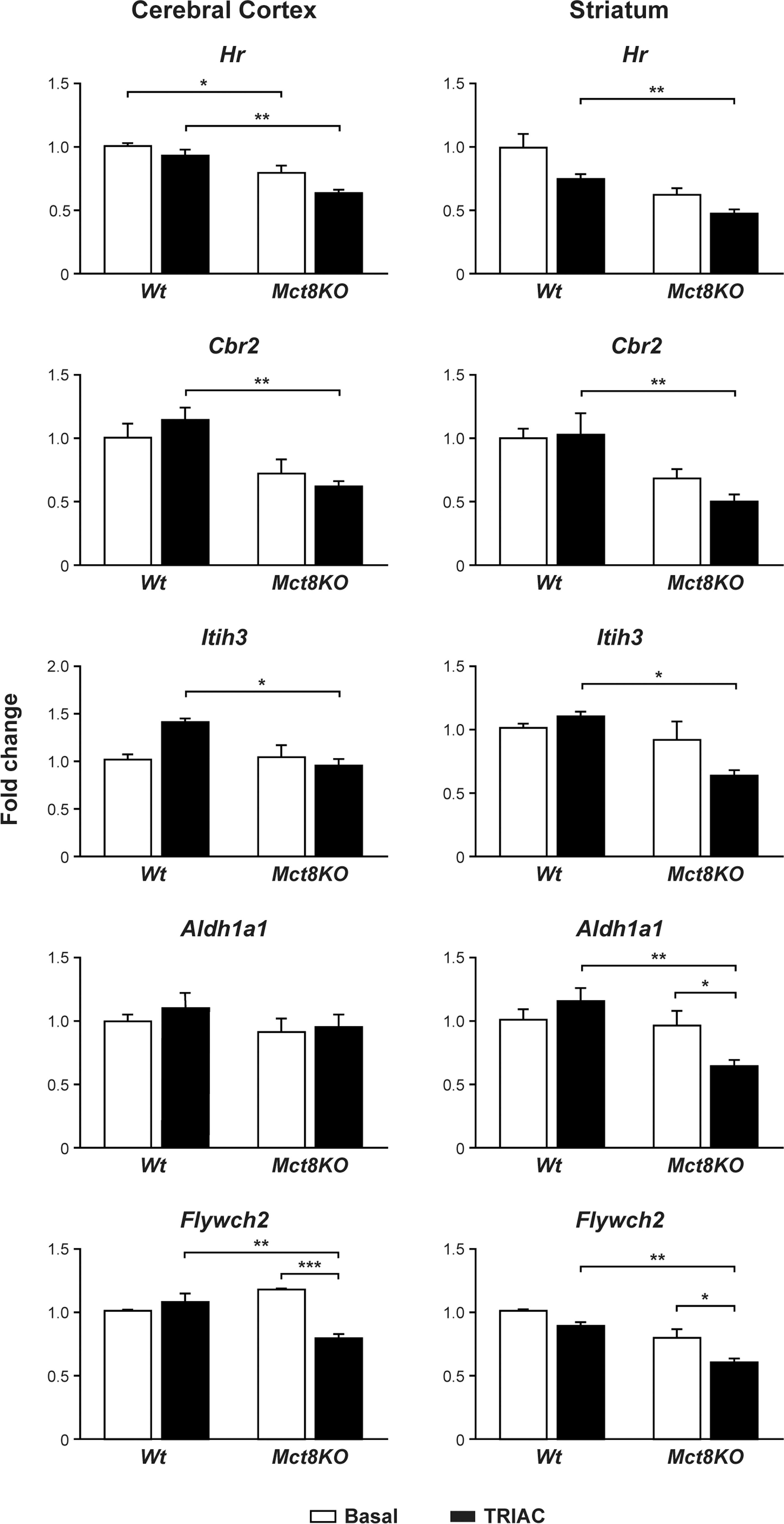
Gene expression analysis of T3-dependent genes (M ± SE) in the cerebral cortex (left panel) and the striatum (right panel) in Wt and Mct8KO mice in basal conditions and after TRIAC treatment. Measurements were obtained by qPCR, and the data are expressed relative to 18S RNA. *p < 0.05, **p < 0.01, and ***p < 0.001 were determined by two-way ANOVA and Bonferroni's post hoc test, the two factors being genotype and treatment. Hr, hairless mRNA; Cbr2, carbonyl reductase mRNA; Itih3, inter-trypsin inhibitor, heavy chain 3 mRNA; Aldh1a1, aldehyde dehydrogenase 1 family, member A1 mRNA; Flywch2, FLYWCH family member 2 mRNA.
The most striking finding in the striatum (Fig. 3, right panels) was the decrease in the expression levels of all the studied T3-dependent genes in the TRIAC-treated Mct8KO animals compared with baseline, although the decrease was only statistically significant for Aldh1a1 and Flywch2.
The ability of TRIAC to reach the brain was evaluated by directly measuring the content of TRIAC in the cerebral cortex and the striatum of Wt and Mct8KO animals. In the cerebral cortex (Fig. 4A, left panel), TRIAC treatment did not increase the TRIAC content in either Wt or Mct8KO mice. The small increase in TRIAC content after treatment was not statistically significant. This was also observed in the striatum (Fig. 4A, right panel) in which TRIAC content did not change at all after treatment.
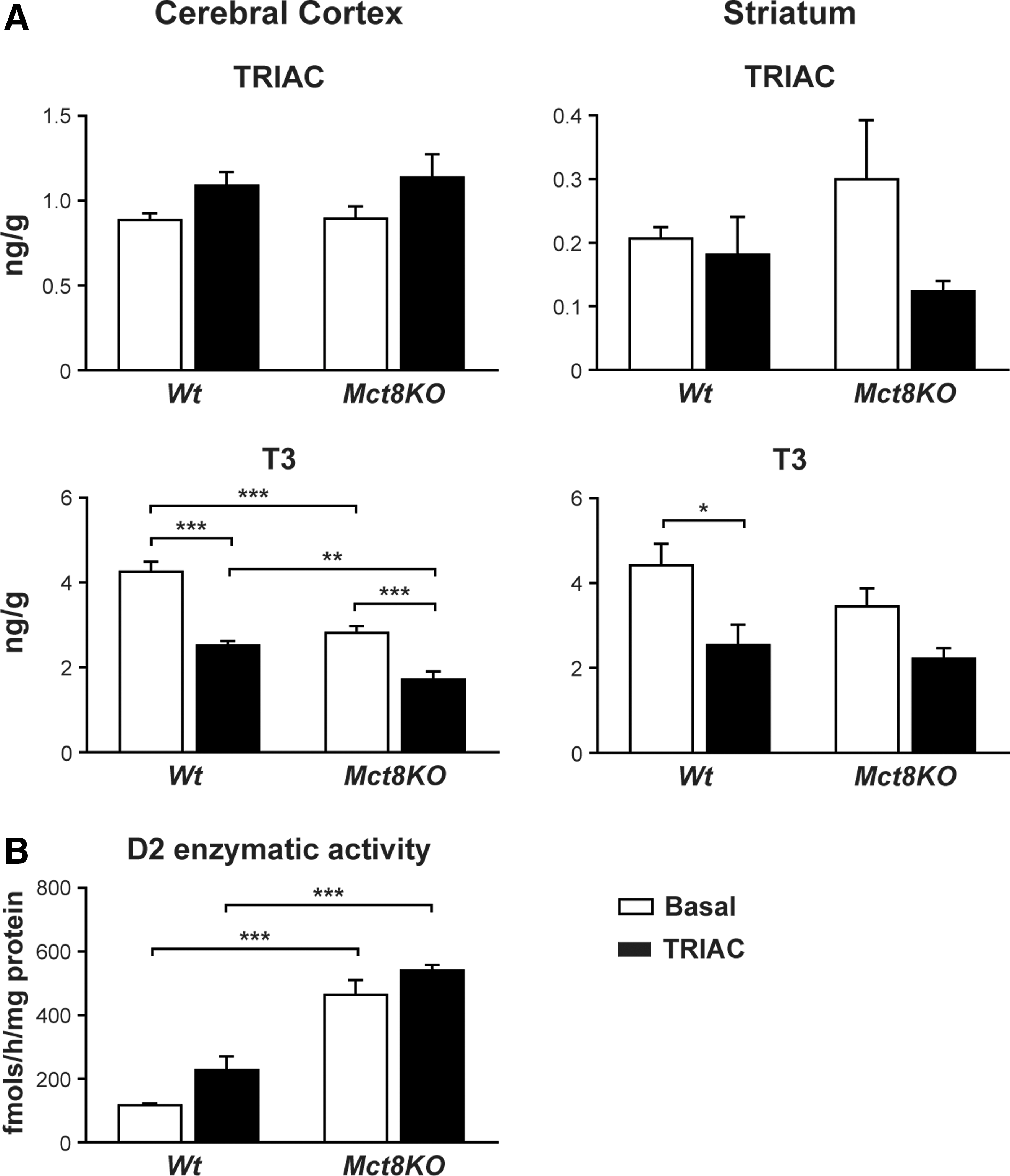
The CNS thyroidal status was assessed by determining the T3 content in the cerebral cortex and the striatum. In the cerebral cortex (Fig. 4A, left panel), the baseline T3 content was lower in Mct8KO than it was in Wt mice, as already reported (18,19). TRIAC treatment induced a decrease in the T3 content of both Wt and Mct8KO animals. The same occurred in the striatum (Fig. 4A, right panel), although the reduction in the Mct8KO mice was not statistically significant.
Measurements of D2 enzymatic activity in the cerebral cortex (Fig. 4B) showed the previously identified elevated D2 activity in Mct8KO animals compared with the Wt mice (18,19), which reflects a compensatory mechanism (34). Nevertheless, D2 activity in Mct8KO animals did not change after treatment, probably due to saturation of D2 activity. In the TRIAC-treated Wt animals, there was a small increase in D2 activity in the cerebral cortex, but it was not statistically significant.
Discussion
The aim of this study was to evaluate the effects of TRIAC in Mct8-deficient mice as a possible candidate for the treatment of patients with MCT8 mutations. TRIAC has many known properties that could serve this purpose. However, it is unclear how therapeutic doses may affect the physiology of Mct8KO mice or if it exerts a function in the brain at the genomic level in vivo, inducing similar neuronal gene responses as T3. Mct8KO mice faithfully replicate the endocrine abnormalities present in patients, and they display alterations in the expression of some T3-dependent genes that enable TRIAC action in the brain to be assessed. Therefore, the goal was to characterize the effects of therapeutic doses of TRIAC in Wt and Mct8KO mice with a special focus on the CNS of juvenile mice.
Wt and Mct8KO littermate mice were treated daily from P21 to P30 with 30 ng of TRIAC/g BW added in the drinking water. After treatment, plasma TRIAC levels greatly increased in both Wt and Mct8KO mice, showing that the dose and administration led to the same effects in both genotypes. Mct8 deficiency does not seem to interfere with the feedback regulation on TSH by TRIAC, since TRIAC treatment caused a drastic decrease in the plasma T4 levels in both the Wt- and Mct8KO-treated animals. Furthermore, taking the plasma T4 levels of the basal Wt animals as a reference, TRIAC-treated Mct8KO animals showed a sixfold reduction. This decrease in plasma T4 levels in TRIAC-treated Mct8KO animals suggests that therapeutic doses of TRIAC lead to severe hypothyroxinemia, which may be potentially harmful unless they are attenuated by the thyromimetic effect of TRIAC.
One of the most relevant findings is that TRIAC treatment restored the elevated plasma levels of T3 in Mct8KO mice, bringing them to normal values without affecting the T3 levels of the similarly treated Wt mice. In this aspect, TRIAC could be a beneficial treatment, as it ameliorates the peripheral tissue hyperthyroidism present in Mct8KO animals.
In the treated Wt mice, the T3 levels in plasma seem to be maintained at the expense of an increased TRIAC-stimulated D1 activity, despite low levels of T4 in plasma. The lower levels of plasma T4 in Mct8KO are not sufficient to maintain plasma T3 levels, despite the increase in D1 activity. This observation is consistent with the similar T4/T3 plasma ratios in the treated Wt and Mct8KO mice (43.5 and 45.2, respectively).
It has been described that TRIAC enters the liver after treatment (33), and TRIAC, as T3, has a direct effect in the liver, since D1 activity and Dio1 expression increased after treatment in the Wt and Mct8KO mice. Therefore, although TRIAC treatment reduced the plasma T3 levels into the reference range in Mct8KO animals, it did not normalize the hyperthyroid state of the liver due to the thyromimetic action of TRIAC.
Consistent with previous studies, TRIAC at this dose does not affect the T3-dependent Serca2a gene expression in the heart. Two separate studies in rodents support an absent effect of TRIAC on the heart when using similar or even higher doses than those used in this study. In these studies, TRIAC failed to increase gene expression and D1 activity, despite entry into the heart (33,43).
Because patients suffering from MCT8 deficiency present with very severe neurological impairments and the therapeutic approaches have so far not been successful in improving this condition, it is essential to determine whether physiological doses of TRIAC are able to reach the brain and exert a thyromimetic action in MCT8 deficiency.
TRIAC has been proven to mediate effects on gene expression in vitro similar to those of T3 (32). In vivo, the established therapeutic dose of TRIAC failed to reproduce this effect in Mct8KO mice. There was no effect in the expression of any of the genes examined, aside from a reduction of expression of a few T3-dependent genes in the cerebral cortex and the striatum, suggesting that therapeutic doses of TRIAC are not sufficient to regulate gene expression differentially in the brain, and it may aggravate the mild hypothyroid situation present in Mct8KO mice. This observation is supported by the direct measures of TRIAC and T3 content in the cerebral cortex and the striatum of Wt and Mct8KO animals at baseline and after TRIAC treatment. In the striatum, there were no differences in the TRIAC content after treatment, in either the Wt or the Mct8KO animals. In the cerebral cortex, there may have been a subtle increase in the TRIAC content in the treated Wt and Mct8KO animals, but this increase was not statistically significant. In terms of T3 content, there was a considerable decrease in T3 after TRIAC treatment in both genotypes in both cerebral regions. These two observations provide direct evidence supporting that this dose of TRIAC is not sufficient to regulate gene expression in brain cells and that it leads to a mild hypothyroid situation. As the primary source of T3 in the brain is generated by local deiodination of plasma T4 into T3 by D2 activity (44), and taking the big reduction in the plasma T4 levels of the treated animals into account (especially in the TRIAC-treated Mct8KO animals), it is not surprising to find a reduction in the T3 content of the brain.
TRIAC treatment did not alter D2 activity in the cerebral cortex of either genotype compared with their basal controls. Previous studies by others with similar doses as the ones used here revealed that TRIAC infusion does not affect D2 cerebral cortex activity of hypothyroid rats (43), showing that TRIAC does not have an effect per se. In the present study, saturation of D2 activity, which is very elevated in Mct8KO mice, could explain this finding.
It is concluded that a therapeutic dose of 30 ng TRIAC/g BW/day in Mct8KO mice restores plasma T3 levels but severely decreases T4 levels. TRIAC induces increased D1 activity and gene expression in the liver, but it does not have any effect on cardiac gene expression. On the one hand, TRIAC has a beneficial effect, reducing the peripheral hyperthyroidism without affecting the heart; on the other hand, TRIAC treatment is not able to normalize the thyrotoxic effect in the liver present in Mct8 deficiency. Thus, it would generate a situation of severe hypothyroxemia that could be harmful, especially for the brain.
It can also be concluded that treatment with established therapeutic doses of TRIAC lead to a threefold increase in plasma TRIAC levels that are insufficient to increase TRIAC levels in the brain and to promote the expression of T3-dependent genes in brain cells. Furthermore, under these conditions, TRIAC treatment generated a situation of brain hypothyroidism with reduced T3 content in the brain.
Previous studies in Mct8/Oatp1c1-double KO mice have demonstrated that high doses of TRIAC (200–400 ng/g) can reach the mouse brain and are able to prevent damage when administered at early stages of development (32). However, the data support that therapeutic doses of TRIAC do not reach the brain of juvenile mice, suggesting that TRIAC treatment for MCT8 deficiency should be considered with caution.
Footnotes
Acknowledgments
We thank Dr. Albert Burger for the TRIAC antibody. We acknowledge Maria Camino de Lucas, Irene Cuevas, and Raquel Arocha for animal care, as well as Javier Pérez for the artwork. We thank Samuel Refetoff for his critical review of the manuscript.
This work was supported by Grants from the Mehuer Foundation (Santiago Grisolía) and the Seville's College of Pharmacists, Ramón Areces Foundation (CIVP16A1805), Spanish Ministry of Economy and Competitiveness (SAF2011-25608, SAF2014-54919-R, SAF 2012-32491), and S2010/BMD-2423 from CAM, and under the frame of E-Rare-2 and the “Centro de Investigación Biomédica en Red de Enfermedades Raras, Instituto de Salud Carlos III” under the frame of the ERA-Net for Research on Rare Diseases. S.B.-L. is recipient of a predoctoral fellowship and contract from the FPI program of the Plan Nacional de I+D+i. The cost of this publication has been paid in part by FEDER funds.
Author Disclosure Statement
The authors have nothing to disclose.