
Abstract
Background:
Recently, the first patients with resistance to thyroid hormone alpha (RTHα) due to inactivating mutations in the thyroid hormone receptor alpha (TRα) were identified. These patients are characterized by growth retardation, variable motor and cognitive defects, macrocephaly, and abnormal thyroid function tests. The objective was to characterize a young girl (18 months old) with a mutation in both TRα1 and TRα2, and to study the effects of early levothyroxine (LT4) treatment.
Methods:
The patient was assessed clinically and biochemically before and during 12 months of LT4 treatment. In addition, the consequences of the mutation for TRα1/2 receptor function were studied in vitro.
Results:
At 18 months of age, the patient presented with axial hypotonia, delayed motor development, severe growth retardation, and abnormally elevated triiodothyronine (T3)/thyroxine (T4) ratios. RTHα was suspected, and concomitantly a c.632A>G/p.D211G missense mutation was identified, affecting both the TRα1 and TRα2 proteins. This mutation was also found in the girl's father. LT4 treatment was started, resulting in a marked improvement of her hypotonia, motor skills, and growth. Functionally, the missense mutation led to decreased transcriptional activity of TRα1, which could be overcome by higher T3 levels in vitro. The mutant TRα1 showed a moderate dominant negative activity on wild type (WT) TRα1. In contrast, WT TRα2 and mutant TRα2 had negligible transcriptional activity and showed no dominant-negative effect over TRα1.
Conclusions:
This report describes the phenotype of a young RTHα patient with a mild TRα mutation before and during early LT4 treatment. Treatment had beneficial effects on her muscle tone, motor development, and growth.
Introduction
T
For more than 20 years, it has been known that heterozygous mutations within three hotspots of the ligand-binding domain (LBD) of TRβ result in resistance to thyroid hormone β (RTHβ), a clinical syndrome characterized by elevated serum TH levels, a non-suppressed thyrotropin (TSH), and a variable phenotype comprising goiter, tachycardia, and increased energy expenditure. Recently, the first patients with RTHα due to mutations in the LBD of TRα1 have been identified. These patients are characterized by growth retardation, variable motor and cognitive defects, macrocephaly, and abnormal thyroid function tests (elevated T3/thyroxine [T4] and T3/reverse T3 [rT3] ratios with normal TSH levels) (6 –12). Recently, Moran et al. identified the first family with a mutation in both TRα1 and TRα2 (13). Patients in this family displayed a similar clinical and biochemical phenotype as patients with a mutation in TRα1 alone, suggesting that the mutation in TRα2 does not contribute to the nature or severity of the phenotype (13,14). Although only a few patients with RTHα have been identified so far, the severity of the phenotype appears to be related to the location and type of the mutation in TRα1. Considering the crucial role of TH during development, RTHα patients may benefit differently from levothyroxine (LT4) treatment, depending on the severity of the mutation and the timing of treatment (6 –9,11 –13).
This study describes the youngest patient identified with RTHα so far. She was diagnosed with a mild mutation in both TRα1 and TRα2, and was treated with LT4 from the age of 18 months. Beneficial effects of treatment on different parameters such as motor development, growth, and thyroid function tests are supported by in vitro data showing that the reduced sensitivity of the mutant TRα1 toward T3 could be overcome by higher T3 concentrations.
Patients, Materials, and Methods
Index patient
Because of suspected central hypothyroidism, the index patient was referred to a pediatric endocrinologist, who conducted extensive thyroid function tests showing low free thyroxine (fT4) and rT3 levels, with a high T3 and a normal TSH concentration. Based on the clinical phenotype and this typical biochemical profile, a mutation in THRA was suspected.
Clinical and biochemical assessment
Written informed consent was obtained from the parents of the index patient. The study was approved by the Medical Ethics Committee of the Amsterdam Medical Center.
From the age of 18 months onwards, the girl underwent detailed physical examinations (growth parameters, head circumference) by the pediatric endocrinologist once every two to three months. In addition, biochemical measurements (T4, fT4, T3, rT3, TSH, thyroglobulin, thyroxine-binding globulin, insulin-like growth factor (IGF)-1, IGFBP-3, sex hormone–binding globulin, ferritin, and complete blood count), imaging (skeletal X-ray), and motor developmental tests (Alberta Infant Motor Scale [AIMS] and the Bayley Scales of Infant and Toddler Development [Bayley III-NL]) (15,16) were performed at specific time points.
Genetic analysis
Sequencing of exons 6-9 of THRA (GenBank: NM_199334.3) was performed as previously described (6). The sequence results were compared to the reference sequence of THRA. To exclude PCR errors, the amplification and sequencing of the exon carrying the mutation was repeated.
Functional characterization of the mutant TRα1 and TRα2: DNA constructs and mutagenesis
The mammalian expression vector pcDNA3-FLAG-TRα1 was used, containing the full-length coding sequence of human TRα1 with a 5′-FLAG tag and optimized Kozak sequence, as previously described (8). The pcDNA3-FLAG-TRα2 expression vector was generated according to the same methods (see Supplementary Table S1 for primers; Supplementary Data are available online at
Mutations in TRα1 and TRα2 were generated by site-directed mutagenesis using the QuickChange II Mutagenesis kit (Agilent Technologies), according to the manufacturer's instructions. The presence of the introduced mutations was confirmed by sequencing.
To study the transcriptional activity of wild type (WT) and mutant TRα1 and TRα2, a construct was used containing a DR + 4 TRE-dependent firefly luciferase reporter and a control renilla luciferase reporter (pdV-L1; gift from Dr. W.S. Simonides, VUMC, NL) (17).
Cell culture and transfection
JEG3 cells were maintained in Dulbecco's modified Eagle's/F12 medium supplemented with 9% fetal bovine serum (FBS) and 100 nM Na2SeO3 at 37°C and 5% CO2. To deplete cells of TH, charcoal-treated FBS (CT-medium) was used instead for 48 h. Cells were transfected at 80–90% confluence with 100 ng pdV-L1 luciferase-renilla reporter construct together with 100 ng WT or mutant TRα1, or a combination of 50 ng WT and 50 ng mutant TRα1 using Xtreme Gene 9 transfection reagent (Roche Diagnostics), according to the manufacturer's protocol. To explore the possible effect of the mutation on TRα2 function, WT TRα1 was co-transfected with WT or mutant TRα2 at different ratios (1:1, 1:10, and 1:50).
Luciferase assays
Transcriptional activity of WT and mutant receptors was determined using the Dual-Glo Luciferase kit (Promega), as previously described (7). The luciferase activity was normalized to the renilla activity to adjust for transfection efficiency. The results are presented as means ± standard error of the mean of at least three independent experiments, carried out in triplicate.
Immunoblotting
To determine expression of WT and mutant TRα1 and TRα2, transfected cells were lysed in ice-cold RIPA buffer supplemented with the Complete Protease Inhibitor cocktail (Roche Diagnostics). The lysates were cleared by centrifugation for 5 min at 700 g, and the protein concentration of the supernatant was determined by bicinchoninic assay (BCA) (Fisher Scientific). Proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked in TBST/5% milk and probed overnight at 4°C with a 1:1000 dilution of the FLAG M2-antibody (F1804; Sigma-Aldrich). Bound antibody was detected with a horseradish peroxidase–conjugated goat anti-mouse antibody (#172-1011; Bio-Rad) and visualized by enhanced chemiluminescence using the Alliance 4.0 Uvitec platform (Uvitec Ltd).
Electrophoretic mobility shift assays
Direct binding of WT and mutant TRα receptors to DNA was tested by electrophoretic mobility shift assay (EMSA), using a double-stranded overlapping oligonucleotide probe labeled with a fill-in reaction using Klenow fragment in the presence of 32P-labeled dCTP (18). The sequences of the oligonucleotides used for the synthesis of the DR + 4 TRE-containing probe are listed in Supplementary Table S1. The 32P-labeled probe was purified using NucAway Spin Colums (Life Technologies). Receptor proteins were transcribed and translated in vitro using the TnT Quick Coupled Transcription/Translation System (Promega), according to the manufacturer's protocol. The binding assays were performed according to previously described methods (19,20). Radioactive spots were visualized by autoradiography.
Statistical analysis
Statistical differences between WT and mutant receptors were calculated using Student's t-test. p-Values of < 0.05 were considered statistically significant.
Results
Case report and clinical assessment of the index patient at baseline
The girl was born at term by vacuum extraction after an uncomplicated pregnancy. Birth weight was 4000 g, and Apgar scores were 5, 7, and 9 after 1, 5, and 10 min, respectively. After an uneventful neonatal period, including a normal neonatal congenital hypothyroidism screening result (T4 62 nmol/L [−1.3 SD], TSH 1 mIU/L), the girl displayed hypotonia and slow motor development, whereupon she was referred to a pediatric physical therapist. At first consultation at the age of 8.5 months, the hypotonia was found to be predominantly axial. Motor developmental testing (15) yielded an AIMS score below the 5th percentile, indicating severe delay in motor development.
At the age of 10.5 months, the girl was referred to a pediatric neurologist for lack of progression in motor development and persisting hypotonia. In the absence of “alarm” signs suggestive of serious neuromuscular disease, and because cognitive development was judged normal, diagnostic testing was postponed and frequent physical therapy (PT) was continued. At the age of 12 months, the girl rolled over from tummy to back and vice versa for the first time, a milestone normally occurring at 9 months (21), but she was still unable to crawl or pull herself up to a standing position, which are characteristic milestones for 12 months (21). Although the AIMS was not routinely performed after the first PT consultation, the physical therapist reported no clear improvement up to the age of 18 months (based on home video fragments filmed between the ages of 17 and 18 months, the physical therapist estimated the AIMS score at that age to be between 20 and 30, still far below the 5th percentile; Fig. 1C).
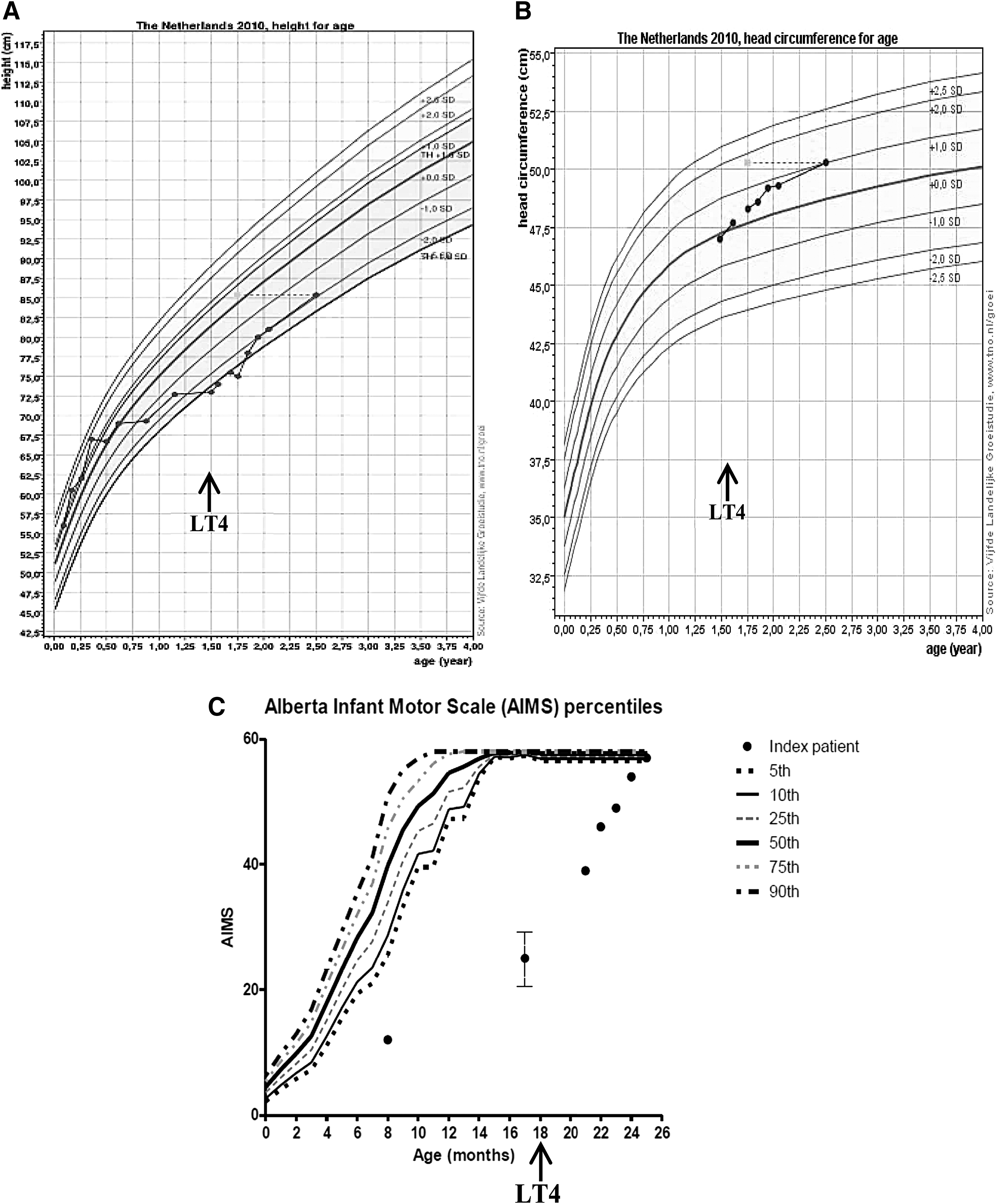
Phenotypic characteristics of the index patient. (
Because of persistent hypotonia, lack of improvement in motor development, and mild speech delay (babbling and the use of only one word), additional investigations were performed around the age of 17 months. A full blood count, blood chemistry (including liver enzymes and creatine kinase), and electromyography were normal. However, repeated laboratory testing revealed fT4 concentrations (9.0–9.8 pmol/L) just below the lower limit of the reference interval (10.0–20.0 pmol/L) in the presence of normal TSH concentrations (2.0–5.6 mIU/L; reference interval 0.48–4.6 mIU/L). At this point, she was referred to a pediatric endocrinologist, at the age of 18 months, for suspicion of central hypothyroidism. The medical history revealed mild constipation from the age of six months onwards, but no other problems. The family history revealed primary hypothyroidism in the maternal grandmother, but no thyroid or pituitary disorders in other family members or in the father's family.
On physical examination, height was 73 cm (−2.77 SD), weight 10.6 kg (+2.11 SD), body mass index (BMI) 19.89 (+2.17 SD), and head circumference 47 cm (0 SD; Fig. 1A and B). The face and nasal bridge were broad, but there was no macroglossia. Skin tags were absent. Neurological examination showed impaired trunk balance with axial hypotonia, and more pronounced hypotonia in the arms than in the legs. Repeated but more extensive thyroid function testing showed low fT4, T4, and rT3, high T3, normal TSH, and increased T3/T4 and T3/rT3 ratios (Table 1), supporting clinical suspicion of a mutation in THRA.
T4, thyroxine; fT4, free thyroxine; T3, triiodothyronine; TSH, thyrotropin; TBG, thyroxine-binding globulin; IGF-1, insulin-like growth factor 1; MCV, mean corpuscular volume.
Genetic assessment
Sequencing of THRA indicated that the index patient and her father were heterozygous for a nucleotide substitution (c.632A>G), resulting in an aspartic acid to glycine substitution at codon 211 (p.D211G) in both TRα1 and TRα2 (Fig. 2). This mutation is not present in public databases.
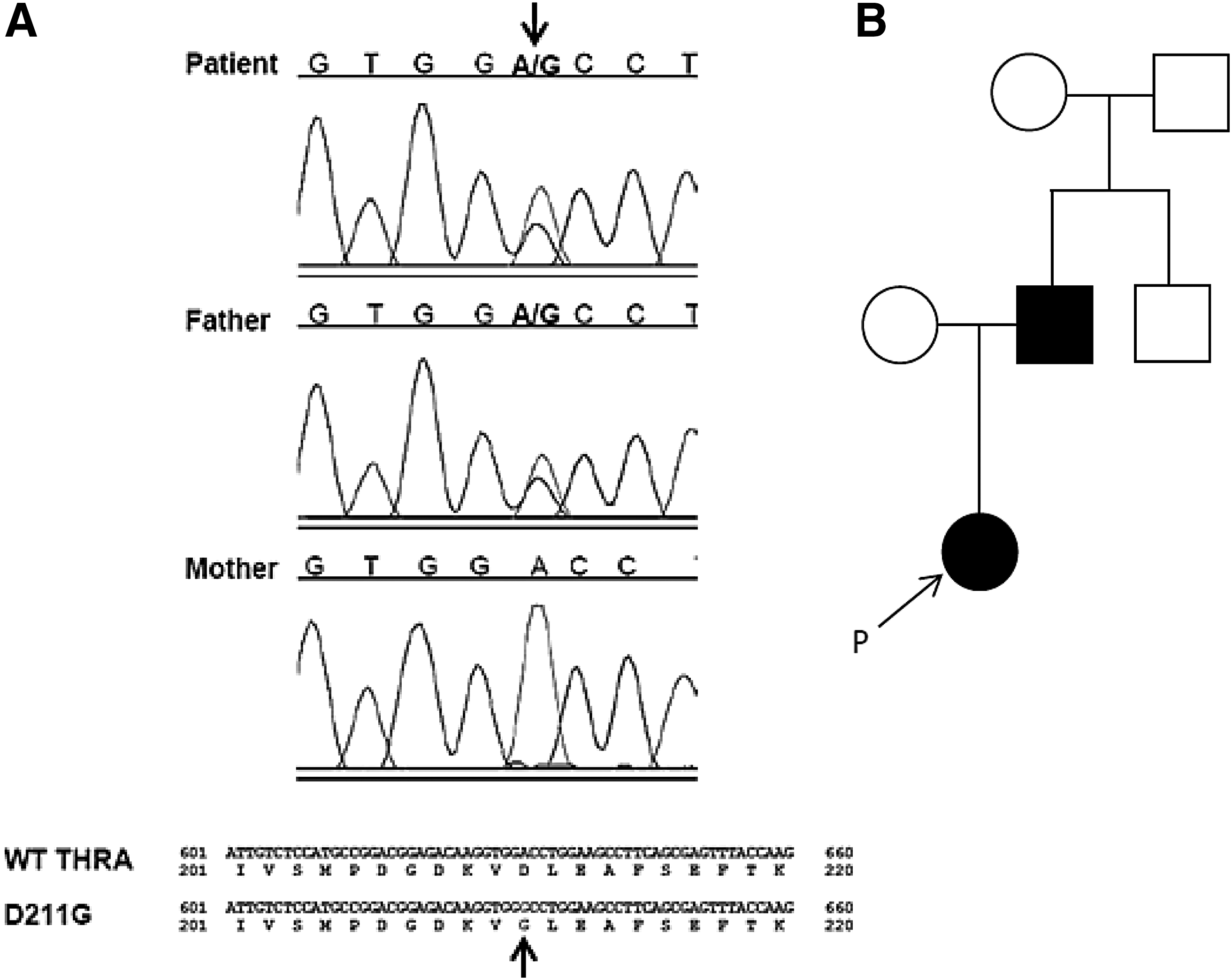
Molecular analysis of THRA in the index patient and her father. (
Clinical assessment of the index patient during LT4 treatment
Because of the presumptive diagnosis of central hypothyroidism, while awaiting the results of THRA mutation analysis, treatment with LT4 was started with a daily dose of 3.5 μg/kg body weight. The dose was gradually increased to a final dose of 62.5 μg (5.25 μg/kg body weight). After six months of LT4 treatment (chronological age of 24 months), the index patient showed clear improvement in motor development: her hypotonia had almost disappeared, and she was able to crawl, sit, and walk independently, albeit with a broad gait. Repeat assessment by the referring pediatric neurologist showed no clear abnormalities. AIMS testing by the same physical therapist yielded a score of 54, corresponding to the 50th percentile at 12 months of age (Fig. 1C). In addition, the parents found their daughter to be livelier, less tired, and her bowel movements had normalized to once daily. Her speech was still slightly delayed (use of a few words, but not yet two-word phrases, the milestone for 24 months) (21). Her length had increased to −1.98 SD and head circumference to +0.76 SD (Fig. 1A and B). Her bone age was delayed (9 months at a chronologic age of 21 months). On LT4 treatment, plasma fT4 normalized, and TSH was suppressed (Table 1).
After one year of LT4 treatment, still at a dose of 62.5 μg/day, the now 30-month-old girl showed further progress in motor development, with Bayley III-NL scores (16) for gross and fine motor skills of 50 and 41, corresponding to developmental ages of 19 and 25 months, respectively. Speech development had also improved: the girl was now able to say two-word phrases (the milestone for 24 months) (21). Formal neurological examination at the age of 32 months revealed only very mild hypotonia, which prevented her from changing from a lying to a sitting position. In order to compensate, she rolled over onto her tummy and pushed herself up to a sitting position. Further assessment showed normal gross and fine motor skills and symmetric reflexes. In addition, her length had increased to −1.92 SD, head circumference to +1.84 SD, and bone age to 21 months (chronological age 30 months), respectively.
Clinical assessment of the girl's father
The father of the patient has the same mutation and has not been treated to date. No detailed data on his childhood development are available, but he reported delayed puberty. At the age of 15 years, he had four epileptic episodes for which he was treated with valproic acid. Furthermore, he suffered from constipation (hard stools two to three times a week). Physical examination showed coarse facies, macrocephaly (+2.5 SD), a short stature (−1.19 SD), and an increased BMI (28.3 kg/m2). Blood pressure (135/85 mmHg) and bone-mineral density (femoral head: Z-score −0.3, T-score −0.5; lumbar spine: Z-score 0.9, T-score 0.9) were normal. In addition, neuropsychological evaluation showed normal performances on memory, language, attention, and executive function tests, together with a normal processing speed. Lastly, his biochemical profile (abnormal T3/T4 and T3/rT3 ratios with a normal TSH and a mild anemia; Table 2) fitted well with the previous patients identified with RTHα (6 –10,13).
Functional analysis of mutant TRα1 and TRα2
To determine the effect of the mutation on TRα1 function, FLAG-tagged WT or D211G-TRα1 was expressed in JEG3 cells, and T3-dependent transcriptional activity was determined. In luciferase assays, the D211G mutant TRα1 showed a reduced apparent affinity for T3 compared with WT TRα1 (EC50 10.1 nM T3 for D211G TRα1 vs. 0.49 nM T3 for WT TRα1; p < 0.001). However, the D211G mutant had a similar maximal transcriptional activity as WT TRα1 (Fig. 3A). When co-expressed, the D211G mutant TRα1 had a moderate dominant-negative effect on WT TRα1 function (Fig. 3A).
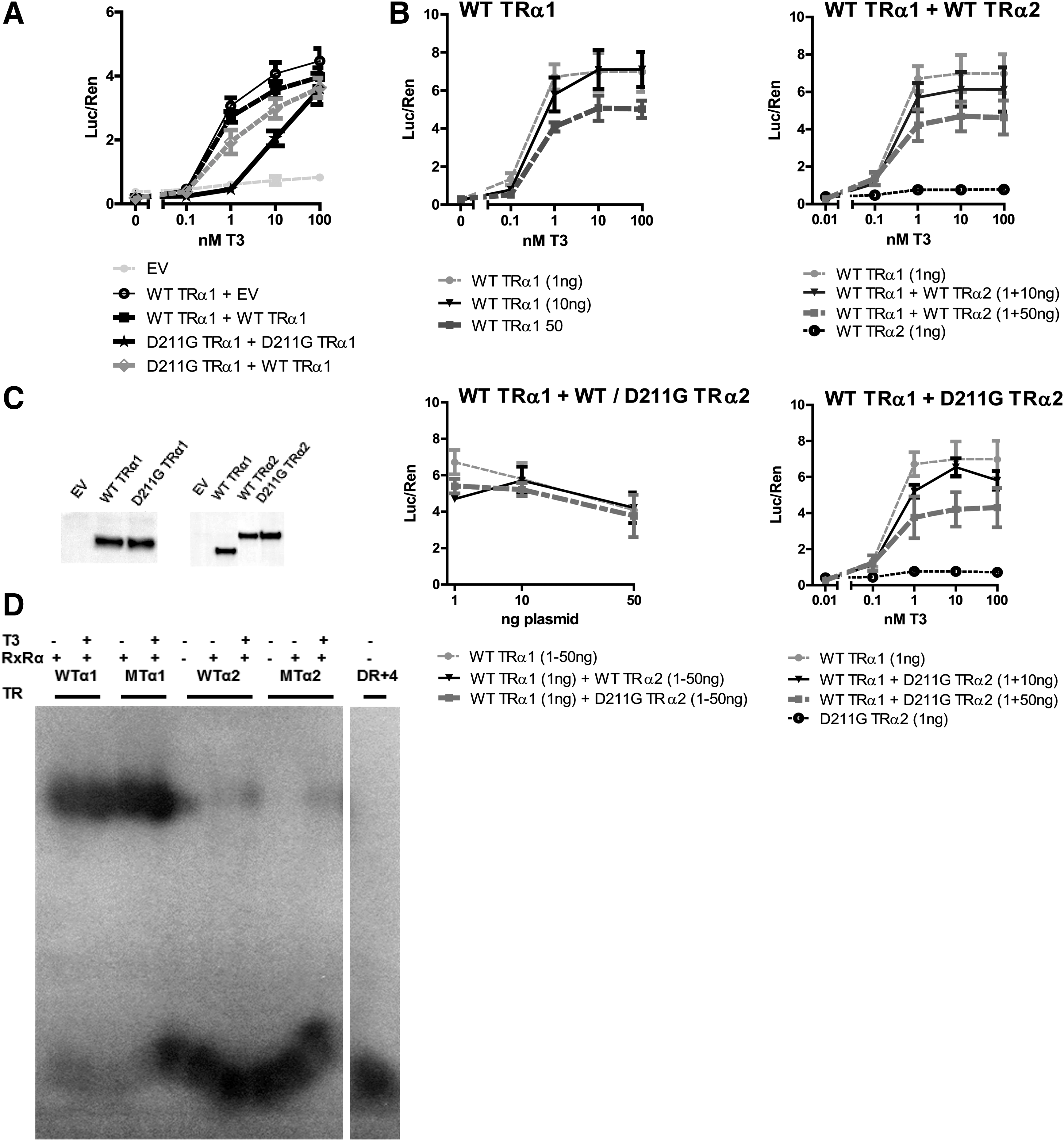
Functional analysis of D211G mutant TRα1 and TRα2. (
Independent of the T3 concentration, WT TRα2 and D211G TRα2 showed no transcriptional activity. Co-transfection of WT TRα1 with increasing amounts of WT or mutant TRα2 (1:1, 1:10, and 1:50) suppressed transcriptional activity, but the same was the case after increasing the amount of WT TRα1 plasmid alone (Fig. 3B). This indicates that both WT and mutant TRα2 exert little or no dominant-negative effect over TRα1.
A similar expression of WT TRα1, WT TRα2, and D211G TRα2 proteins was confirmed by immunoblotting lysates for FLAG (Fig. 3C).
In addition, the binding capacity of WT and mutant TRα1 and TRα2 to a 32P-labeled DR + 4 TRE probe was tested by EMSA in the absence or presence of RXRα and T3. WT and mutant TRα1 bound similarly to the TRE as heterodimers with RXRα independent of T3, whereas no binding of WT TRα2 or mutant TRα2 to the probe was detected under any of the conditions tested (Fig. 3D). In the absence of RXRα, both WT and mutant TRα1 still bound to the TRE, but the binding was less pronounced (data not shown).
Overall, these results suggest that D211G TRα1 has a reduced affinity for T3, while DNA binding and expression levels are similar to WT TRα1. At high concentrations of T3, maximal transcriptional activity of the mutant is the same as for WT TRα1. In contrast to TRα1, both WT TRα2 and D211G TRα2 show no transcriptional activity nor binding to the DR + 4 TRE.
Discussion
This study describes the effects of early LT4 treatment in a very young girl with a mild missense mutation in TRα1/2. Beneficial effects during treatment on her muscle tone, motor development, and growth are reported. This is in line with the in vitro data showing that the reduced transcriptional activity of the mutant receptor could be overcome by higher T3 concentrations, and that D211G TRα1 has a moderate dominant negative effect on WT TRα1. The D211G mutation in TRα2 does not influence the function of WT TRα1 or TRα2, and does not seem to contribute to the phenotype of the patient.
Although the index patient had some clinical characteristics associated with hypothyroidism (growth retardation, and a broad face and nasal bridge), her hypotonia and delay in motor development were the prime reasons for referral and consultation. Because of presumed central hypothyroidism, based on normal TSH with low-normal fT4 levels, LT4 treatment was started at the age of 18 months. LT4 treatment had a beneficial effect on the girl's development: her muscle tone improved, and her motor development accelerated. In addition, during treatment, she showed catch-up growth and a normalization of her fT4 and rT3 levels. The suppressed TSH concentrations upon LT4 treatment are in line with previous reports (7) and indicate an intact HPT-axis feedback, which is predominantly regulated via TRβ2 (22). The in vitro data support the developmental benefit of LT4 treatment, since transcriptional activity of the mutant TRα1 could be restored in vitro by higher T3 concentrations.
The fact that the girl's father reached adulthood without major complications associated with the TRα1 D211G mutation supports the relatively mild character of this mutation with a decreased but not absent affinity for T3. The difference in severity between the developmental phenotype of the father and the daughter is remarkable, but similar to RTHβ in which the severity of the phenotype also varies among different subjects carrying the same mutation (23). The reasons for this variability are not yet fully understood, but may result from genetic variability in other genes, such as cofactors involved in TH action.
Anemia is a frequent characteristic in RTHα, and was only present in the father of the index patient. Usually, it concerns a normocytic normochromic anemia. However, in three cases, the mean corpuscular volume was raised (8,9,14). Several studies in humans have indicated an association between hypothyroidism and anemia (24,25). In addition, data from animal models point toward an important role for TRα in erythropoiesis (26 –29). The molecular mechanisms underlying the anemia in RTHα patients remain to be elucidated.
The phenotype of the girl shows similarities with that of heterozygous TRα1-R384C mutant mice, having a mutant receptor with a 10-fold reduced affinity for T3 (30). Affected animals show delayed postnatal development, together with locomotor dysfunctions. Interestingly, treatment with supra-physiological TH doses in the postnatal period, but not in adulthood, substantially improved motor function (31). These TRα1-R384C mutant mice also suffer from growth retardation, which is overcome in adulthood, since they reach a near-normal size (30,32).
The phenotype of the index patient and her father resemble the clinical characteristics of previously described RTHα patients with a mutation in TRα1 alone (6 –11). The D211G mutation in TRα2 does not seem to contribute to this phenotype, which is in line with what has been described in three patients with the A263V mutation, also affecting TRα1 and TRα2 (12,13). The current findings are also in line with the phenotype of the TRα2KO mice, since the phenotypic changes in these mice are most likely not attributable to the loss of TRα2, but rather to a compensatory overexpression of TRα1 in that particular mouse model (33). In contrast, a recently described patient (14) with a N359Y mutation in TRα1/2 showed a distinct clinical phenotype consisting of severe bone malformation, chronic diarrhea, macrocytic anemia, and hypercalcemia. To date, it remains unclear whether this phenotype is the result of the mutation in THRA alone. The current in vitro results support a lack of effect of the mutation in TRα2, since WT and D211G TRα2 showed no transcriptional activity both in the absence or presence of T3. Co-transfection of WT TRα1 with large amounts of WT or D211G mutant TRα2 suppressed the function of WT TRα1 in the same way as when the concentration of WT TRα1 plasmid alone was increased. This suggests that the suppressive effect is caused by an increased amount of transfected plasmid and not by a dominant-negative effect of the TRα2 receptors toward WT TRα1.
The lack of effect of WT or mutant TRα2 on the transcriptional activity of TRα1 is explained by the observation that TRα2 does not bind to the TRE (DR + 4) used in the luciferase assays, independent of the absence or presence of RXRα. Under the conditions of the experiments, competition for binding to the TRE is therefore absent and so is a suppressive effect of TRα2 on TRα1 function. These findings are supported by the study by Liu et al., describing that the possible dominant-negative effect of TRα2 on TRα1 is not attributable to competitive binding to TREs or inadequate formation of heterodimers with RXRα (34).
The fact that the girl's father has RTHα with a relatively mild phenotype, which was detected only because of severe developmental delay of his daughter, supports the hypothesis that the incidence of RTHα could be much higher than currently estimated. Suggestively, the RTHα incidence could correspond to the incidence of RTHβ (1:40,000) (23), given the strong homology between the TRα1 and TRβ1 receptors. Moreover, although so far only a few mutations in TRα have been reported, they seem to cluster in a few hotspots similar to the pattern of TRβ mutations (35). Lastly, the paternal inheritance of the TRα mutation in this family, and maternal and paternal inheritance of TRα mutations in three other families (6,7,10,13), suggests that transmission of TRα mutations from parents to offspring is less impaired in humans than in mice (36).
This case study illustrates that RTHα patients with mild mutations, especially when diagnosed at a young age, are likely to benefit from LT4 treatment. This is supported by a previous case (13), as well as by studies in TRα mutant mice (30,37).
Since the index patient was initially suspected of having central hypothyroidism, a low threshold to measure T3 (and rT3) is advocated in the workup of children with unexplained growth retardation or developmental delay who have a low or low-normal fT4 in combination with a normal TSH.
Footnotes
Acknowledgments
We thank Leontien van der Aa and Erna Langius for their contribution to the clinical assessment of the index patient.
A.L.M. van Gucht, M.E. Meima, and R.P. Peeters are supported by a Zon-MWTOP Grant (number 91212044) and an Erasmus MC MRACE Grant.
Author Disclosure Statement
The authors have nothing to disclose.