
Abstract
Background:
Congenital hypothyroidism due to thyroid dysgenesis (CHTD) is a disorder with a prevalence of 1/4000 live births, the cause of which remains unknown. The most common diagnostic category is thyroid ectopy, which occurs in up to 80% of CHTD cases. CHTD is predominantly not inherited and has a high discordance rate (>92%) between monozygotic (MZ) twins. The sporadic nature of CHTD might be explained by somatic events such as autosomal monoallelic expression (AME), given that genes expressed in a monoallelic way are more vulnerable to otherwise benign monoallelict genetic or epigenetic mutations.
Objective:
The aim of this study was to search for complete (90%) AME in normal and dysgenetic thyroid tissues.
Methods:
Aggregated analysis of whole-exome and bulk RNA sequencing was performed on two ectopic thyroids, four normal thyroids, and the human thyroid cell line Nthy-ori.
Results:
A median of 5062 (range 2081–5270) genes per sample showed sufficient numbers of heterozygous single nucleotide polymorphisms to be informative. The median monoallelic expression represented 22 (range 16–32) of the informative genes for each thyroid sample. Examples of genes displaying AME are FCGBP, ZNF331, USP10, BCLAF1, and some HLA genes; these genes are involved in epithelial–mesenchymal transition, cell migration, cancer, and immunity.
Conclusions:
AME may account for the high discordance rate observed between MZ twins and for the sporadic nature of CHTD. These findings also have implications for other pathologies, including cancers and autoimmune disorders of the thyroid.
Introduction
C
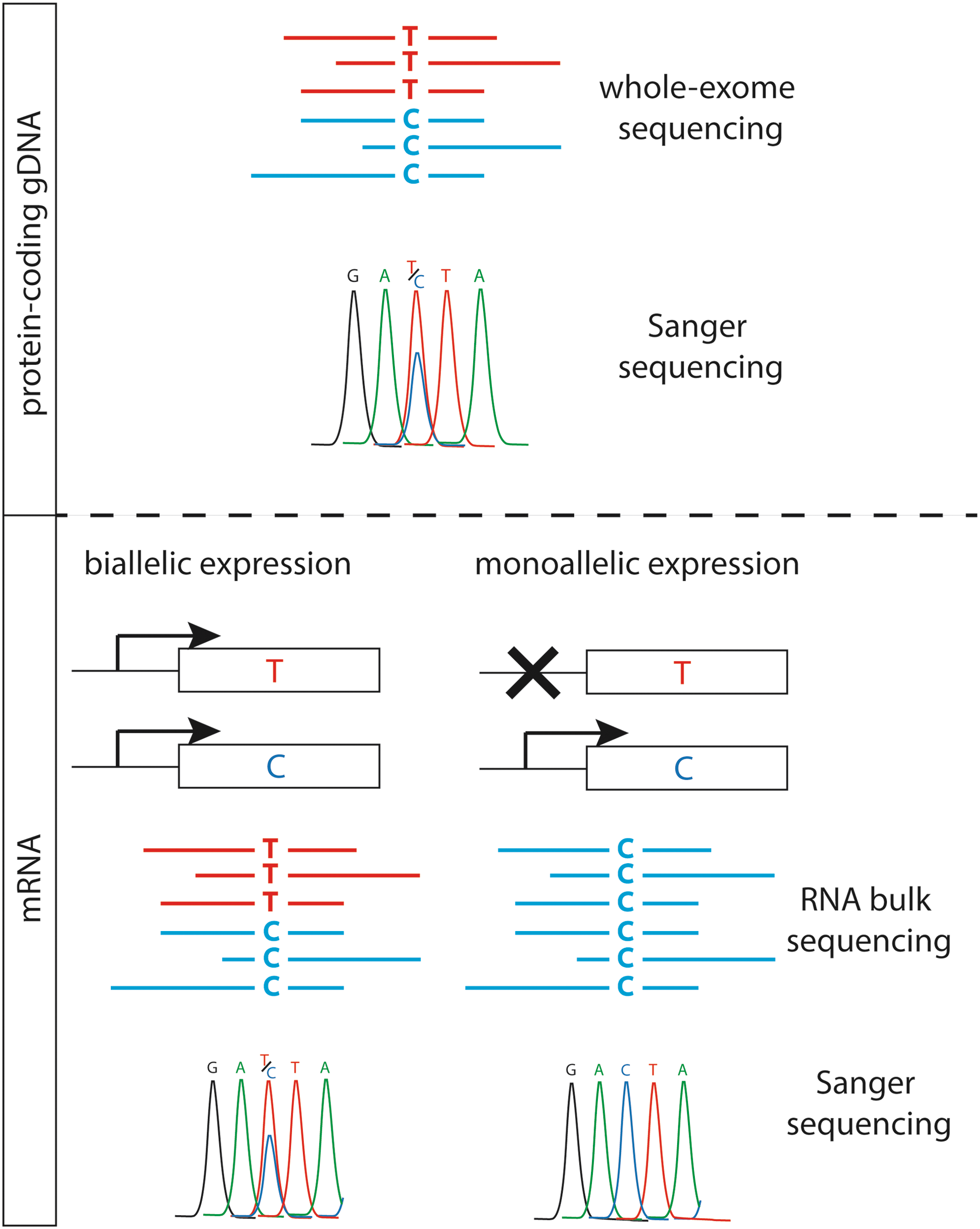
Schematic representation of the autosomal monoallelic expression assessed by whole-exome and bulk RNA sequencing. Color images available online at
Patients and Methods
Patients and tissue samples
Flash-frozen samples of ectopic thyroid tissue were obtained that had been removed from two girls (aged 10 and 8 years) due to local symptoms (dysphagia). These two girls were treated with levothyroxine and had normal thyroid hormones levels before surgery (Supplementary Table S1; Supplementary Data are available online at
DNA and RNA extraction from tissue samples
DNA was extracted using a pureLink genomic DNA Mini kit (Life Technologies, Burlington, Canada). RNA was extracted using a miniRNeasy kit (Qiagen, Toronto, Canada).
WES and bulk RNA sequencing
Sequencing was performed in the McGill University and Genome Quebec Innovation Centre. WES was performed using the Agilent 50 Mb SureSelect exon capture library, followed by 2 × 100 nt paired-end sequencing on the Illumina HiSeq 2000 instrument as described previously (7,15). Paired-end and strand-specific RNA-sequencing was performed in five controls and two case samples (see sample characterization in Supplementary Table S1).
Analysis of bulk RNA sequencing
Analyses were carried out with aligned RNA-sequencing data, where alignment was based on the established TopHat 2 pipeline. To assess allelic expression, high-quality variant calls of exome sequencing data (generated separately), the variant call files were used for local genotype imputation/phasing (in RefSeq genes) using IMPUTE2 software with 1000 Genomes reference haplotypes (Phase I, vs3) in two main steps: pre-phasing and imputation. Variants with an r
2 value of <0.8 and phase genotypes were filtered out using SHAPEIT2 with 1000 Genomes reference haplotypes. In the case of singleton single nucleotide polymorphisms (SNPs), an attempt was made to phase based on common heterozygous sites in paired-end reads. Allele-specific analyses for transcriptome utilized a careful realignment process to phased genomes, coupled with combined tests for allelic abundance of each transcript as recently described (van de Geijn, BioRxiv, 2014;
Combined analysis of WES and bulk RNA sequencing
To define monoallelic expression, variants heterozygous in WES and nominally or seemingly homozygous in RNA sequencing data were identified. To generate a list of testable candidate variants, the number of genes was reduced using a hierarchization pipeline for each sample (Fig. 2). First, to select genotypes from the exome data, the software SAMtools was used (16), and the variants were filtered through the following criteria: artefacts outside the exome were filtered out, number of reads in exome data (≥10), and genotype quality score (QUAL <50). Only good-quality variant calls (genotypes) were used in the latter analysis. Second, to assess which genes showed complete AME, RNA-sequencing data of the informative genes were filtered through the following criteria: percentage of reads in RNA (either <10% or >90% of the altered or reference reads, respectively), inclusion of genes with two or more variants (to eliminate false positives), and exclusion of imprinted genes (as listed in

Breakdown of the experimental protocol, including bioinformatics pipeline and filtering of whole-exome and bulk RNA sequencing data.
Comparison of WES data from matched DNA leukocytes and ectopic thyroids
For WES, 100–120-fold median coverage of targeted exonic regions was typically obtained using the Agilent SureSelect plus Illumina HiSeq technologies. Following read alignment, variant calling, and gene annotation by the bioinformatics analysis pipeline, each exome typically yielded >100,000 candidate variants, of which approximately 25,000 are either in or immediately adjacent to coding exons. To assess for somatic mutations in ectopic thyroids, the genotype of all variants called in the RNA sequencing data (after filtrations) was verified by comparing the number (and percentage) of reads between thyroids and leukocytes.
Results
Gene expression clustering of thyroid tissues
To assess the quality of the thyroid tissue samples with respect to contamination with other tissue types, an unsupervised hierarchical clustering of the RNA sequencing data was performed. As compared to other human cells (fibroblasts, monocytes, and B- and T-lymphocytes), the gene expression profiles derived from all thyroid tissues clustered together and were distinct from other human cells in a cluster dendrogram (Supplementary Fig. S1, left side branch). These results indicate that the thyroid RNA samples were clean.
Exome sequencing, RNA sequencing, and autosomal monoallelic expression in thyroid tissue
To determine whether thyroid tissue shows autosomal monoallelic expression (AME), RNA-sequencing combined with exome sequencing was performed on seven different thyroid samples: 9078 genes with good-quality exonic SNPs were covered by WES. To exclude non-informative variants (homozygous variants) and false-positive variants, a hierarchization pipeline was used for each sample (Fig. 2). First, a median of 5062 (range 2081–5270) genes per sample showed sufficient numbers of heterozygous SNPs to be informative. Then, the median complete AME represented 22 (range 16–32) of the informative genes for each thyroid sample, and AME represented a small fraction of the informative genes (0.46%; range 0.31–0.96%). Numbers of specific (non-recurrent) genes in ectopic thyroids are comparable to that of control tissues. Next, validation with Sanger sequencing was performed on cDNA of each sample and confirmed AME for the following genes: ZNF331, CTSF, ADRB2, USP10, BCLAF1, and FCGBP (Fig. 3). Finally, 30 genes (e.g., some HLA region genes) were recurrently (in two or more samples) subject to AME, and seven of these genes are located on chromosome 6 (Table 1). Only a few genes belong to the same pathways (assessed in KEGG pathway database;

Monoallelic expression in autosomal genes: confirmation by Sanger sequencing of monoallelic expression in (
Genes located on chromosome 6 are noted in bold italic. Validated genes are identified with an asterisk.
Absence of somatic mutations in the two ectopic thyroid samples
To assess whether an early post-zygotic mutation was observed in ectopic thyroids, the exome of the matched leukocytes and the thyroid of cases 1 and 2 (leukocytes and thyroid compared for each case separately) were sequenced and compared. Although these exome comparisons initially yielded a large number of candidate somatic variants, after bioinformatics quality filtering and Sanger resequencing of some candidates, no thyroid-specific mutations were observed; in particular, there were no tissue-specific variants in any of the genes currently known to be involved in thyroid development and/or associated with thyroid dysgenesis. As expected, all genes showing AME were found to have heterozygous variants in both leukocytes and thyroid tissue from the same patient.
Discussion
In humans, genes are generally biallelically expressed, with each parental homologous gene contributing to mRNA expression. However, in some cases, only one copy is expressed, which defines monoallelic expression. There are three known general mechanisms causing monoallelic expression: (i) imprinting, in which imprinted genes are expressed preferentially or exclusively from either the paternal or maternal allele (sometimes with tissue specificity); (ii) random but clonally inherited X-chromosome inactivation in female cells; and (iii) non-predetermined (“random”) AME.
In fact, the first genes discovered to be subject to AME were those encoding immunoglobulins and T-cell receptors, by yet a different mechanism involving DNA rearrangement (17,18). However, as far as we know, this mechanism is specific to these gene families. In 1994, AME was reported in olfactory receptors genes (a family of >1000 genes), which suggested that autosomal AME might affect additional autosomal genes (19). Eventually, in 2007, a genome-wide survey analyzing clonal human blood cell lines revealed widespread AME across the autosomes (8). Over the last years, two forms of autosomal AME have been defined (20). On one hand, fixed AME implies that the allele-specific expression is conserved in daughter cells after division. Thus, fixed AME is mitotically conserved and can be studied in cell lines and tissues (or subtissue sections). On the other hand, dynamic AME is observed only during a window of time, is not mitotically transmitted, and can be studied only at the single-cell level. The present study therefore assessed fixed AME in thyroid tissues and in a thyroid cell line. Of note, given that (i) some classes of genes seem to be more prone to AME (8,12,21) and (ii) herein a recurrence of AME for a median of 15 genes per thyroid sample was observed, AME probably reflects a yet unknown tissue-specific genetic and epigenetic phenomenon (22), but is unlikely to be a completely “random” event. The somatic interplay between genetic and epigenetic mechanisms that control establishment and maintenance of widespread AME remains to be discovered (9).
In thyroid tissue, monoallelic expression of TPO has been shown in cases of thyroid dyshormonogenesis (23,24), and it has been hypothesized that PAX8 monoallelic expression in the thyroid could explain a familial case of congenital hypothyroidism associated with athyreosis (25). Moreover, RNA sequencing analysis recently showed a widespread biased allele-specific expression (an attenuated form of AME) in thyroid tissue (10). This prompted the search for autosomal genes displaying AME in thyroid tissues by using paired exome and RNA sequencing of seven different thyroid tissues.
The results show AME for a median of 22 genes per sample in ectopic and orthotopic thyroid glands, as well as in the Nthy-ori cell line. These findings confirm that genes in the thyroid are subject to AME. The number of autosomal genes subject to AME per tissue was relatively modest. This can be explained by the design of this study and by some technical limitations. First, only genes with an almost exclusive monoallelic expression were selected: the expressed allele is the almost exclusively transcriptionally active allele (>90% of reads in RNA sequencing), while the other one is transcriptionally silent. Second, a significant expression level in the thyroid (a minimum of 10 reads) was required in order to call a gene subject to AME (Fig. 2). Therefore, the results may represent only the extreme end of a continuum, beginning with allele-biased expression (or allele-specific expression (10)) and ending with high-confidence genes subject to complete monoallelic expression (20). Of note, the median of 0.46% of genes subject to AME was consistent with the 0.7% of AME observed in neural progenitor cells (26). The number of informative genes was relatively low, but this was expected because most of the SNPs are present in non-coding regions of the genome, which are not covered by exome and bulk RNA sequencing. For example, the median number of 5062 informative genes per sample was consistent with the genome-wide study of AME conducted in B-cells by Gimelbrant et al., which showed about 3900 informative genes per sample (8).
CHTD is predominantly a sporadic (random) event, and ectopic thyroid, the main form of CHTD, results from an arrest in the migration of the precursor cells. By exome sequencing, it was previously shown that MZ twins discordant for thyroid dysgenesis do not differ in the DNA extracted from nucleated blood cells (7), making early post-zygotic somatic mutation a less likely cause of CHTD. Similarly, no somatic epimutations were observed in the ectopic thyroids (6). The absence of any validated thyroid-specific somatic mutations in the present study is consistent with all these previous findings. Taken together, these results imply that early post-zygotic coding region mutations, or altered epigenetic markings, are unlikely in CHTD. In contrast, the extensive AME observed in all thyroid samples assessed in the present study might well account for the discordance between MZ twins and the sporadicity observed in CHTD. Indeed, genes subject to AME might contribute to cellular (clonal) diversity (8) and are more vulnerable to otherwise benign heterozygous genetic or epigenetic mutations (27).
Only three recurrent genes are common in the two ectopic glands (CDC27, CTBP2, and MST1L), and they do not belong to similar cellular pathways (as assessed by the KEGG pathway database;
Stochastic variable gene expression among genetically identical cells has been implicated in processes including determination of cell fates (33,34) and the development of genetic diseases (11). Reduction of the stochastic expression noise, either by haploinsufficiency or by AME, increases the likelihood of impaired gene function and disease (11). A dynamic AME and transcription dynamic has been recently uncovered in embryonic cells (35). It is therefore likely that stochastic dynamic AME has even more fundamental implications for the penetrance and severity of congenital disorders, including CHTD (35). Somatic variation in the proportion of cells with normal biallelic expression compared to cell with AME of a pool of genes (9) might also lead to the broad range of phenotypes observed in CHTD, ranging from athyreosis to thyroid ectopy and thyroid hemiagenesis.
In conclusion, this study is the first to show clearly that complete AME is observed for groups of genes in ectopic and orthotopic thyroids. These findings, together with the reported tendency of surface proteins to be AME prone, suggest that a pathogenic variant expressed monoallelically in ectopic thyroid tissue might account for the almost universal discordance between MZ twins for this malformation. This latter point remains speculative, as this study is limited by the low number of ectopic thyroids, which are difficult to obtain because patients with thyroid ectopy almost never require surgery to remove the ectopic gland. This would imply that CHTD is a genetically driven stochastic event, in which morphogenic genes might interfere with cell motion, local physical chemoattractant gradients, and even local physical constraints, as suggested six decades ago by Turing (36,37). It is likely that the role of AME in sporadic congenital disorders is greater than currently appreciated (27).
Footnotes
Acknowledgments
We thank the patients and their parents for their cooperation. This work was supported by a grant from the Canadian Institutes of Health Research (MOP-130390 to J.D.). Research in pediatric thyroid diseases at Centre Hospitalier Universitaire (CHU) Sainte-Justine is supported by the Girafonds/Fondation du CHU Sainte-Justine (to J.D. and G.V.V.).
Author Disclosure Statement
The authors have nothing to disclose.