
Abstract
Background:
The thyroid hormone–inactivating enzyme type 3 deiodinase (D3) is induced during hypertrophic and ischemic cardiomyopathy, leading to a state of local cardiac hypothyroidism. Whether D3 induction occurs in dilated cardiomyopathy is unknown.
Methods:
This study characterized changes in cardiac D3 and thyroid hormone signaling in a transgenic model of progressive dilated cardiomyopathy (TG9 mice).
Results:
Cardiac D3 was dramatically induced 15-fold during the progression of dilated cardiomyopathy in TG9 mice. This D3 induction localized to cardiomyocytes and was associated with a decrease in myocardial thyroid hormone signaling.
Conclusions:
Cardiac D3 is induced in a mouse model of dilated cardiomyopathy, indicating that D3 induction may be a general response to diverse forms of cardiomyopathy.
Introduction
H
Delineating the role of T3 in the failing heart also relies on an emerging understanding of the role of thyroid hormone activation and inactivation within specific tissues, catalyzed by the iodothyronine deiodinase family of selenoenyzmes. The thyroid gland primarily secretes the prohormone T4, which is converted in peripheral tissues into the active hormone T3 by types 1 and 2 deiodinase (D1 and D2) (16,17). In contrast, type 3 deiodinase (D3) inactivates both T4 and T3 by inner-ring deiodination to reverse T3 (rT3) and 3,5-diiodo-L-thyronine, respectively (18). Thus, all tissues are exposed to uniform levels of circulating thyroid hormone, but the differential expression of activating (D1 and D2) and inactivating (D3) deiodinases in individual tissues allows local regulation of thyroid hormone action.
Although there is negligible deiodinase expression in healthy cardiomyocytes, the induction of cardiac D3 has been demonstrated in animal models of both hypertrophic (19 –22) and ischemic cardiomyopathy (23,24). These rodent studies show that failure-induced cardiac D3 dramatically decreases local T3 signaling (20,23,25), and a recent report of positive D3 immunostaining in the cardiomyocytes of patients with ischemic cardiomyopathy indicates that this physiology also extends to humans (26). Studies of D3 knockout mice show that heterozygous animals with a paternally inherited null allele have markedly reduced cardiac D3 expression due to tissue-specific Dio3 gene imprinting. These mice develop restrictive cardiomyopathy that is characterized by myocardial fibrosis and decreased tolerance to isoproterenol-induced cardiac hypertrophy (22). While the confounder of D3 insufficiency in other paternally imprinted tissues complicates the interpretation of this model, these findings support a crucial role for D3 in cardiac function and remodeling and justify further study of D3 induction in cardiac disease.
This study investigated the hypothesis that D3 is induced in the dilated form of cardiomyopathy by studying a transgenic murine model of dilated cardiomyopathy, the TG9 mouse (27). Marked induction of D3 mRNA and activity was observed in the hearts of TG9 mice as failure advanced, confirming that myocardial D3 is induced in dilated cardiomyopathy. D3 expression was localized to cardiomyocytes by immunostaining and associated with decreased expression of T3-responsive genes. These findings support the concept that D3 induction is a general response of the cardiomyocyte to diverse forms of heart failure.
Materials and Methods
Animals
Transgenic overexpression of Cre recombinase in cardiomyocytes, as driven by the α-myosin heavy chain (αMHC) promoter, causes dilated cardiomyopathy and progressive congestive heart failure in the TG9 mouse (27). Based on the consistent finding of heart failure in seven independent founder animals, heart failure in this model is presumed to be caused by the high level of myocardial Cre expression and not by disruption of other genes by insertion of the Cre construct (27). All experiments used male TG9 mice or wild-type mice of the same strain (FVB). Tissues were flash frozen for enzyme/RNA analysis or fixed in formalin for histology. Experiments were approved by the Boston Children's Hospital Institutional Animal Care and Use Committee.
Deiodination assays and serum thyroid hormone measurements
Frozen tissues were assayed for D3 activity, as previously described (28). Serum total T4 and T3 were measured, as previously described, using a modified Coat-a-Count radioimmunoassay (Siemens) and T3 charcoal uptake to correct for serum binding and express results as fT4 index (fT4I) and free T3 index (fT3I) (29,30).
Immunohistochemistry
D3 immunohistochemistry was performed using a polyclonal rabbit anti-D3 antibody (D3-18; 1:50 dilution), as previously described (31,32). Isotype-negative controls were performed.
Gene expression analysis
Total RNA was extracted from tissues using Trizol (Ambion) and reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). Quantification of mRNA was performed by the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad), as previously described (33). The housekeeping gene GAPDH was used as an internal control. Primer sequences are available upon request.
Statistics
Data are presented as means ± standard error of the mean. Analyses were performed by two-factor analysis of variance using a test of age × group interaction to assess the differential change with age between groups. p-Values <0.05 were considered significant.
Results
D3 is induced in the cardiomyocytes of TG9 mice with dilated cardiomyopathy
TG9 mice develop ventricular dilatation that is first detectable at about six weeks of age. Left ventricular dilatation and contractile function then worsen over a highly consistent time course, with death occurring at around 12 weeks. By 11 weeks of age, TG9 mice showed signs of severe congestive heart failure, including decreased activity level, labored breathing, and poor peripheral perfusion (27). The hearts of the TG9 mice at 11 weeks demonstrated pathologic features of heart failure, including biventricular dilation (Fig. 1A and B) and occasional atrial thrombi. Histologic examination of the TG9 hearts at 11 weeks revealed chronic changes of moderate-to-severe myocyte hypertrophy when compared with either TG9 mice at 4 weeks or to nontransgenic mice at 4 or 11 weeks, and also showed occasional myocyte cell death with evidence of healing (Fig. 1C and D).

Type 3 deiodinase (D3) is induced in the failing hearts of TG9 mice. Compared with nontransgenic controls (
Like humans with chronic heart failure, TG9 mice developed increased heart width (p < 0.001; Fig. 1E). The development of heart failure in TG9 mice was accompanied by marked induction of cardiac D3 expression, with a 17-fold increase in Dio3 mRNA compared to age-matched, nontransgenic mice (p = 0.002; Fig. 1F). This degree of upregulation was comparable to that of atrial natriuretic factor (p = 0.002; Fig. 1G), an established biomarker of heart failure and ventricular strain. Cardiac D3 activity was also increased 15-fold (p = 0.001; Fig. 2A). This D3 induction was tissue specific; there was no significant increase in the brains (p = 0.24; Fig. 2B) of TG9 mice. As expected from prior reports of hepatic D3 induction in critically ill mice (33) or humans (34), D3 activity was increased in the livers (p = 0.007; Fig. 2C) of TG9 mice during failure. Immunohistochemistry localized D3 expression in the failing hearts of TG9 mice to cardiomyocytes (Fig. 2D–E).
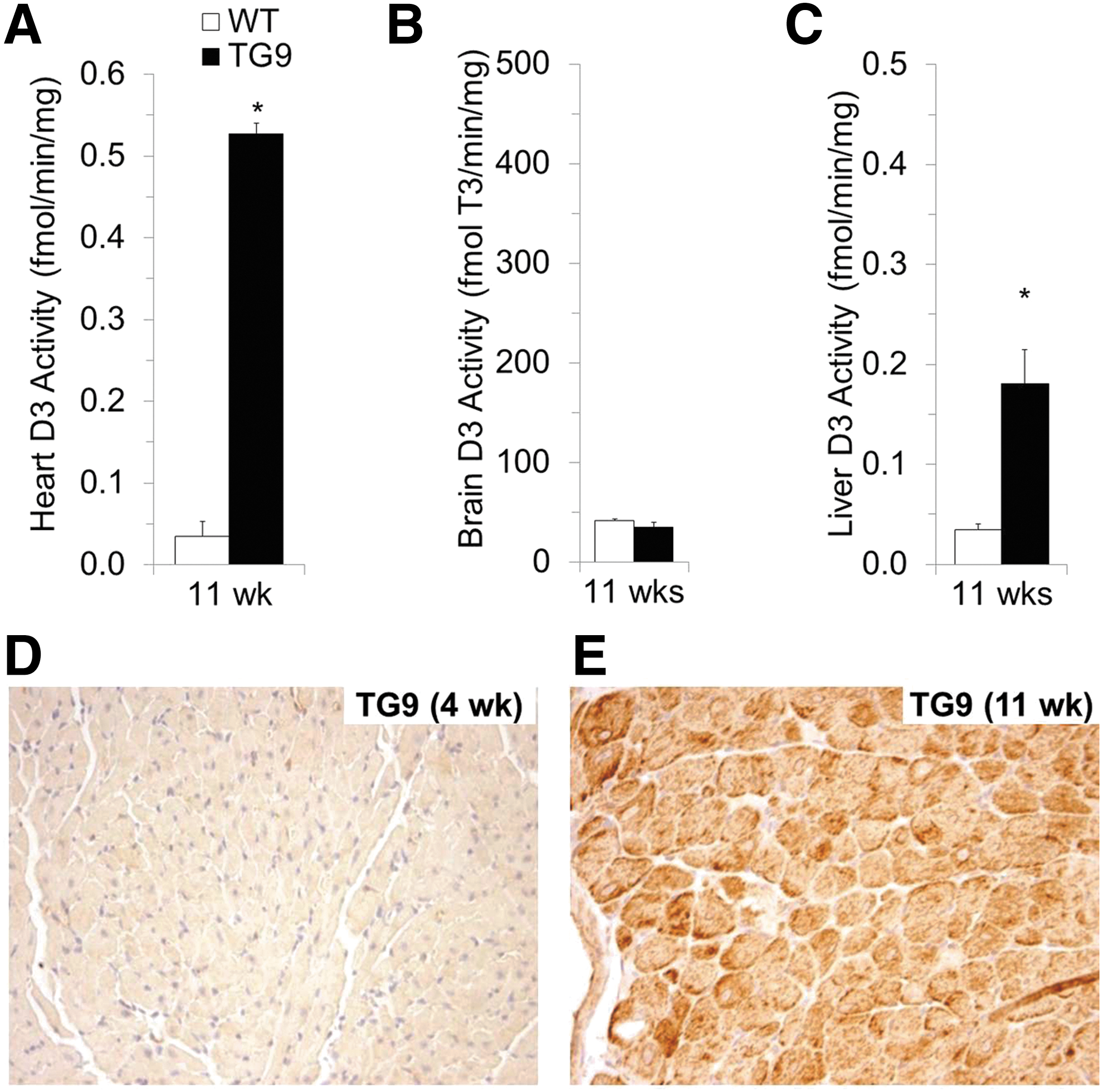
D3 activity induced by dilated cardiomyopathy is tissue specific and localizes to cardiomyocytes. Paralleling the increase in cardiac Dio3 mRNA, D3 activity is markedly induced in the failing hearts of TG9 mice (
Dilated cardiomyopathy in TG9 mice leads to reductions in systemic thyroid hormone status and T3-responsive cardiac gene expression
Recapitulating the suppression of systemic thyroid status observed in chronic heart failure and other severe nonthyroidal illness (4 –8,35,36), the progression of heart failure in TG9 mice was associated with significant decreases in circulating thyroid hormones, with a 68% fall in the serum fT4I and a 71% fall in the serum fT3I (p < 0.001; Fig. 3A and B).
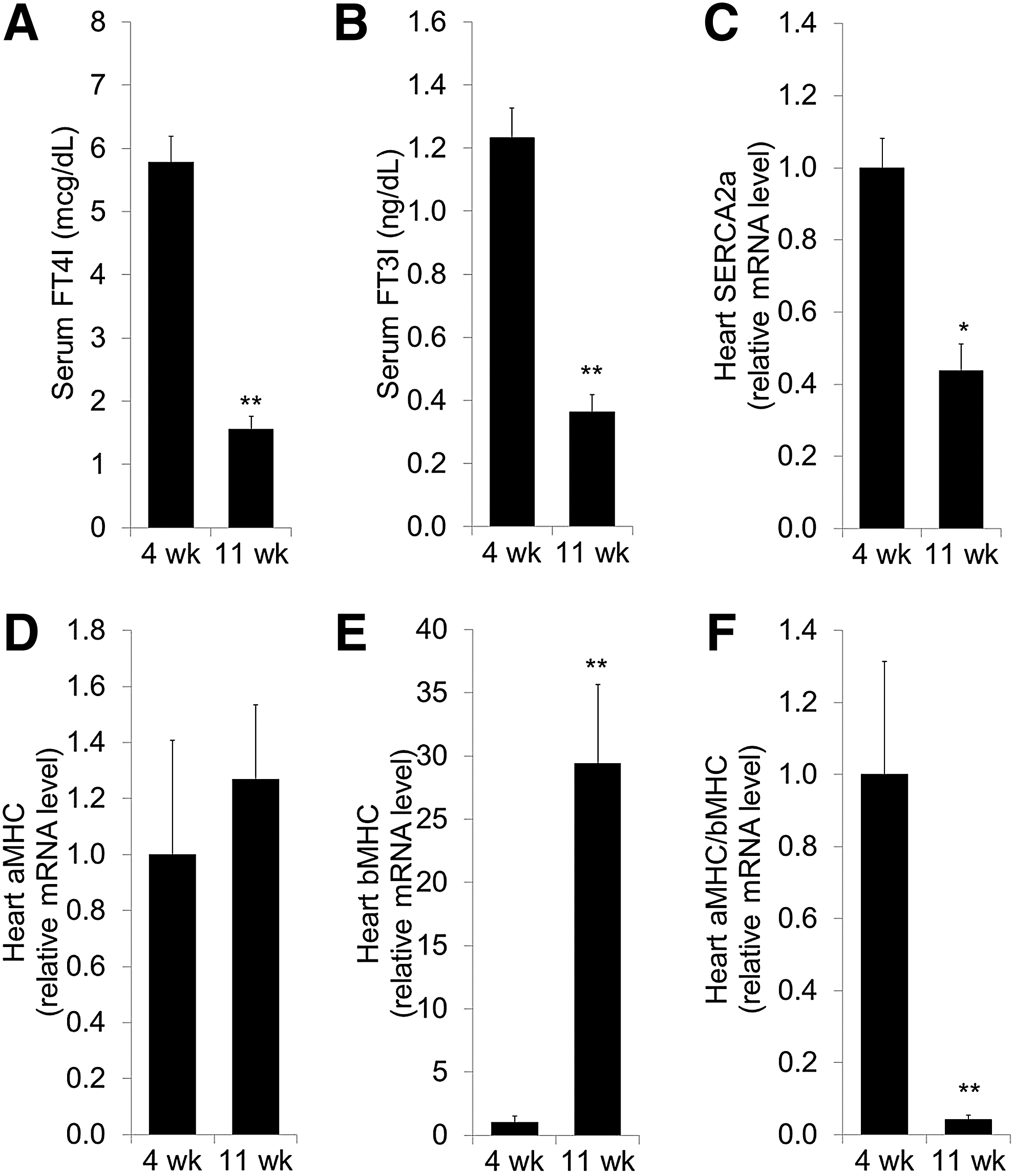
Dilated cardiomyopathy in TG9 mice is associated with reductions in systemic thyroid hormone status and in the cardiac expression of thyroid hormone–responsive genes. Dilated cardiomyopathy is associated with a decrease in serum free thyroxine index (
To determine if the reduction in systemic thyroid status and the induction of cardiomyocyte D3 in TG9 mice was sufficient to decrease local thyroid hormone signaling in the failing heart, the expression of cardiac genes that are regulated by T3 was measured (Fig. 3C–F). SERCA2a (Atp2a2) and αMHC (Myh6) are positively regulated by thyroid hormone, whereas β-myosin heavy chain (βMHC, Myh7) is negatively regulated (37). Since αMHC and βMHC are regulated in opposite directions, the ratio of αMHC/βMHC expression is a sensitive marker of cardiac thyroid hormone signaling. Consistent with cardiac-specific hypothyroidism, there was a 56% decrease in SERCA2a mRNA (p = 0.004; Fig. 3C) and a dramatic 95% reduction in the αMHC/βMHC ratio (p < 0.001; Fig. 3F) in D3-expressing TG9 hearts. The fall in αMHC/βMHC ratio was mediated primarily by increased expression of βMHC (Fig. 3E) with little change in αMHC (Fig. 3D), similar to the expression pattern associated with myocardial D3 induction in a model of ischemic cardiomyopathy (23).
Discussion
Thyroid hormone has profound effects on cardiac function, inducing changes in heart rate, myofibril structure, intracellular calcium release and reuptake, and electrical activity (37,38). These effects have long been recognized in patients with severe primary hypothyroidism who manifest decreases in heart rate, cardiac output, and myocardial efficiency that are fully reversible with thyroid hormone replacement. Illustrating that thyroid status is also regulated locally, animal studies have shown that deiodinase expression in cardiomyocytes is sufficient to alter tissue-specific T3 signaling in the heart, even in the absence of changes in systemic thyroid function. This concept was first established by studies of D2 transgenic mice that overexpress human D2 in their hearts. Due to increased local activation of T4 to T3, these mice have tachycardia and increased cardiac metabolic rate (39 –41). Illustrating the impact of local deiodination on cardiac pathophysiology, these D2 transgenic mice display altered tolerance to doxorubicin-induced cardiotoxicity (41) and aortic banding (21). Because human cardiomyocytes express negligible D2, the direct relevance of these findings to patients remains unclear. However, these experiments are robust proof of the concept that cardiomyocyte deiodination is a potent mechanism to regulate T3-dependent function and injury tolerance in the heart.
In recent years, high D3 expression has been discovered in rodent models of hypertrophic and ischemic cardiomyopathy (23,25) and also in human hearts with ischemic cardiomyopathy (26). This cardiac D3 induction is sufficient to produce anatomically specific hypothyroidism (25), and one study of mice with lifelong D3 deficiency supports that myocardial D3 induction plays a protective role in hypertrophic heart failure (22). Whether D3 induction occurs in other forms of heart failure was unknown. The present study demonstrates for the first time that cardiac D3 activity is dramatically increased in a murine model of dilated cardiomyopathy. The absolute level of cardiac D3 activity induced in failing TG9 hearts is similar to that measured in other rodent models of ischemic (23,24) or hypertrophic (25) cardiomyopathy, and the localization of D3 to cardiomyocytes in TG9 hearts matches the pattern reported in ischemic cardiomyopathy (20,23,26).
Considering that dilated cardiomyopathy is pathophysiologically and histologically distinct from the hypertrophic and ischemic forms of cardiomyopathy (42,43), the data suggest that cardiac D3 induction is a general, fundamental response to myocardial injury and strain. Although the mechanisms underlying cardiomyocyte D3 induction are not fully defined, one possible stimulus shared among various forms of myocardial injury is local hypoxia, which leads to the activation of hypoxia-inducible factors (HIFs). It has previously been shown that HIF-1α is a transcriptional stimulator of the Dio3 gene and mediates D3 induction and local cardiac hypothyroidism in a rat model of hypertrophic heart failure (25). In fact, cardiomyocyte hypoxia alone is sufficient to induce D3 in isolated primary rat cardiomyocytes (25), and studies of monocrotaline-induced right ventricular hypertrophy show robust D3 induction in hypertrophic cardiomyocytes prior to the onset of heart failure (19). The demonstration of increased HIF-1α expression in human hearts with chronic hypertrophic (44), ischemic (26), or dilated cardiomyopathy (45) suggests that HIF-1α activation may be a common mechanism for D3 induction across diverse forms of heart failure. Additional patient studies to define the patterns of D3 induction in human cardiomyopathy are warranted.
In summary, this study used a novel mouse model to show that cardiomyocyte D3 is induced during dilated cardiomyopathy. This D3 induction was associated with decreased systemic thyroid status and the altered expression of thyroid hormone responsive cardiac genes. Further study is justified to identify the key targets of thyroid hormone signaling in chronic heart failure and to define the specific role of D3 in cardiac disease. The recent precedents of conditional knockout mice with hepatocyte-specific (33) or muscle stem cell–specific (46) D3 deficiency, which have revealed important functional roles of D3 in nonthyroidal illness and tissue regeneration, warrant the creation of cardiomyocyte-specific knockout mice to dissect D3's functional roles in various forms of heart failure and the specific contribution of cardiomyocyte D3 to the low T3 syndrome.
Footnotes
Acknowledgments
The authors thank Dr. P. Reed Larsen for his helpful insights and comments on this manuscript and Dr. Henry A. Feldman for assistance with statistical analyses. This work was supported by the Pan-Mass Challenge Fund, the William F. Milton Foundation Award, a Research Grant from the American Thyroid Association, a donation from the Murray Family, and the National Institutes of Health grants DK076099, HL105857, DK044128, DK007699, and HD052896.
Author Disclosure Statement
The authors have nothing to disclose.