
Abstract
Six patients are described with bi-allelic DUOX2 variants and widely variable phenotypes. Patient 1 is an infant with a compressive hypothyroid goiter causing respiratory distress, which was promptly alleviated by levothyroxine (LT4). He was a compound heterozygote for DUOX2 variants, including a novel deletion of 540 base pairs. Patients 2 and 3 are siblings with the same compound heterozygous mutations of DUOX2, yet one had overt hypothyroidism at 14 months and the other lifelong euthyroidism. Patient 4 is a compound heterozygote individual and has mild persistent congenital hypothyroidism; his sister (patient 5) only had a borderline thyrotropin elevation at newborn screening, consistent with homozygous DUOX2 variants with a mild impact on enzyme activity. Their euthyroid mother (patient 6) is a compound heterozygote for the same DUOX2 mutations as her son. Targeted exome sequencing did not reveal any relevant modifiers. It is concluded that (i) prompt LT4 replacement in infants with respiratory distress due to a hypothyroid goiter makes surgery unnecessary; and (ii) the clinical expression of DUOX2 deficiency varies widely between individuals and over time, justifying periodic reevaluation of the need for LT4 replacement.
Introduction
In 2002, Moreno et al. reported that bi-allelic mutations of DUOX2 lead to permanent congenital hypothyroidism, while mono-allelic mutations lead to transient congenital hypothyroidism (1). Since then, genotype–phenotype correlations have been found to be more complex. In addition, mutations in the maturation factor DUOXA2, which is required for the insertion of DUOX2 at the apical membrane, have also been described (2). Six patients harboring bi-allelic DUOX2 variants with a wide spectrum of clinical presentations are described.
Patients
The biochemical parameters of the patients at newborn screening, diagnosis, and reevaluation are summarized in Table 1. Figure 1 shows the DUOX2 variants found in the patients and their family members, with biochemical parameters at presentation.
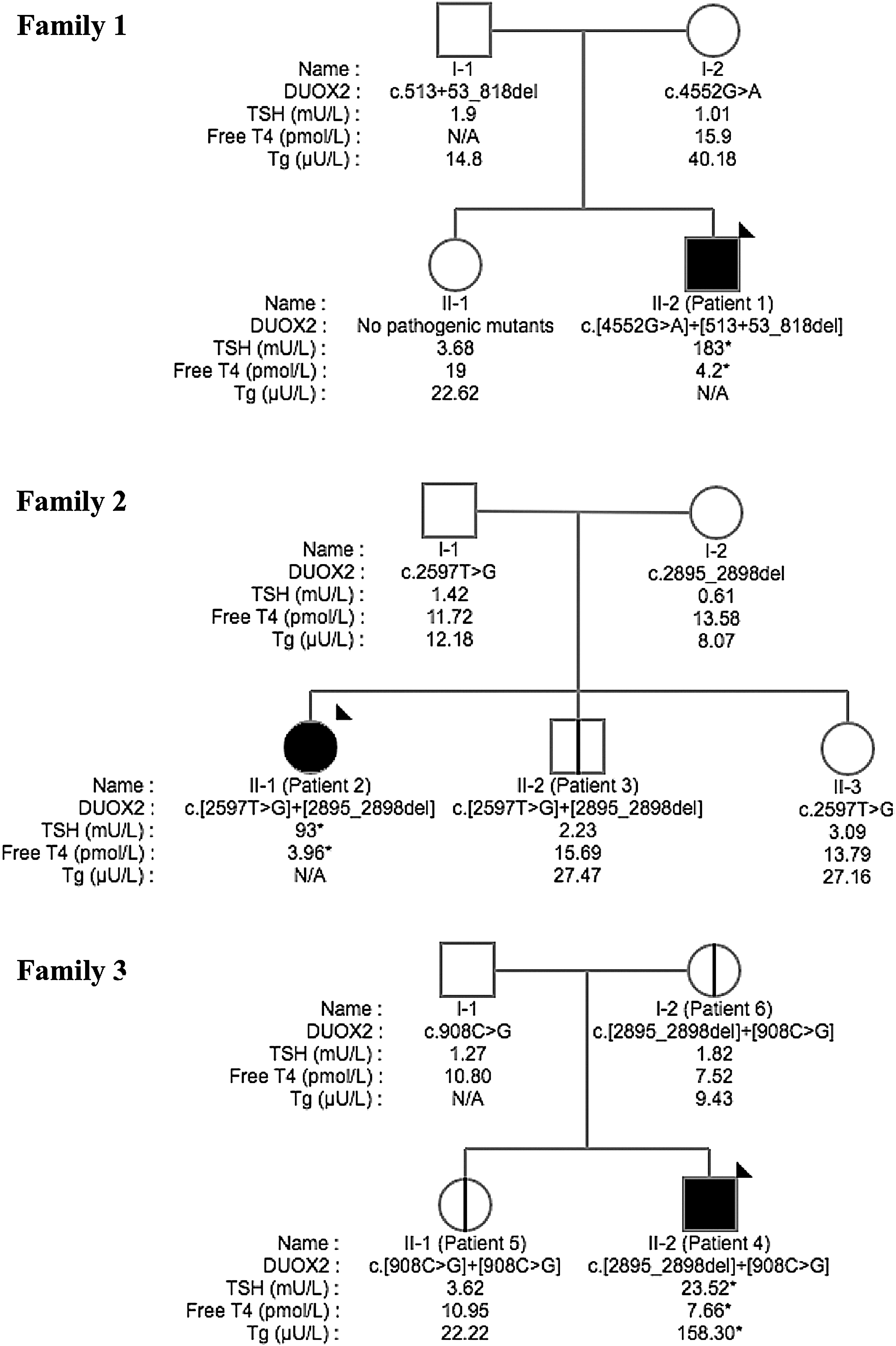
Pedigrees, with the proband in black and indicated by an arrow head, and the results of thyroid function tests at presentation indicated below each symbol, with abnormal values indicated by an asterisk.
Biochemical Parameters of Six Patients from Three Families with Biallelic DUOX2 Mutations
Values outside of the reference range for age are indicated in bold.
TSH, thyrotropin; T4, thyroxine.
Patient 1, the second son of non-consanguineous French-Canadian parents, was born at 40 weeks' gestation by spontaneous vaginal delivery after an uneventful pregnancy. Birth weight was 3940 g. Newborn screening for congenital hypothyroidism was negative. At the age of 13 weeks, growth progressed along the 50th percentile. However, laborious breathing prompted admission. On examination, there was respiratory distress without cyanosis. On nasopharyngo-laryngoscopy, bulging of the right pharyngeal wall was found; the epiglottis deviated from the midline, and the mobility of the right vocal cord was decreased by 50%. The day after admission, a head and neck magnetic resonance imaging (MRI) scan showed a massively enlarged thyroid (Fig. 2). Urgent surgical decompression was considered. Overt hypothyroidism with goiter established the diagnosis of dyshormonogenesis, and normal linear growth and bone maturation confirmed its rapid onset. Oral levothyroxine (LT4) was initiated at 50 μg/day for two days and then continued at 75 μg/day. Breathing became normal by day 3. Serum free thyroxine (fT4) on that day was 17.8 pmol/L (reference range 8.0–18.0 pmol/L), confirming adequate absorption. On continuing replacement, growth and psychomotor development have been normal. His older brother and parents are euthyroid.

Transversal view of the neck magnetic resonance imaging scan of patient 1: the anterior–posterior diameter of both thyroid lobes reached 24 mm with a 50 mm cranio-caudal length. At the superior pole, both lobes were particularly bulky and joined on the midline, causing the right pharyngeal bulging noted on endoscopy (see text). Tracheal compression was particularly severe just above and 1 cm below the glottis, with a sagittal diameter of 2.3 mm (arrows).
Patient 2 is the first of three children born to a French-Canadian mother and a father of Italian descent. Her initial presentation has already been described (3). At the age of 15 years, she had been euthyroid on 37.5 μg/day LT4 for 10 years, but treatment withdrawal resulted in mild hypothyroidism. Patient 3, the younger brother of patient 2, had normal newborn screening, psychomotor development, and growth. At the age of 13 years, thyroid function tests were normal, and at 21 years of age, he remained clinically euthyroid. The perinatal history was negative for iodine overload in both. At ages 15 and 13 years, respectively, urinary iodine was in fact higher in the hypothyroid patient than in her brother (248 vs. 116 μg/L). The mother, father, and younger sister are euthyroid.
Patient 4 was born to healthy non-consanguineous French-Canadian parents. The pregnancy was unremarkable, and labor was induced at 41 weeks' gestation. Birth weight was 3965 g. Newborn screening for congenital hypothyroidism was borderline on day 3. Repeat newborn screening results on day 18 were again borderline. Therefore, the baby was referred on day 25. Mild hypothyroidism with a high serum thyroglobulin was confirmed. A technetium-99m scan demonstrated a slightly enlarged orthotopic thyroid with increased uptake. LT4 replacement was started. Psychomotor development and growth progressed normally. At the age of 32 months, LT4 was withdrawn for six weeks, after which mild hyperthyrotropinemia was documented and LT4 resumed. Patient 5 is the older sister of patient 4. Her thyrotropin (TSH) at newborn screening for congenital hypothyroidism was 14 mIU/L at two days of age. Growth, psychomotor development, and thyroid function tests at 13 years of age were normal. The father and mother (patient 6) are euthyroid. The mother was born before newborn screening was implemented in Quebec. She had normal serum TSH at 33 years of age (postpartum) and again at the age of 45 years.
Additional Studies
Details of additional studies are shown in Figure 1 and Supplementary Table S1. Written informed consent for genetic testing was obtained from all of the children's legal guardians. In patients 1, 2, and 4, exome sequencing targeting 14 genes involved in congenital hypothyroidism (DUOXA2, DUOX1, DUOX2, FOXE1, GNAS, IYD, NKX2-1, NKX2-5, PAX8, SLC26A4, SLC5A5, TG, TPO, and TSHR) was performed, and the DUOX2 variants identified were confirmed by Sanger sequencing. All parents and the sister of patients 2 and 3, as well as the sister of patient 4, were tested for DUOX2 variants.
Patient 1 harbored a previously described missense mutation (p.G1518S) known to abolish DUOX2 activity (2), as well as a novel 541 base-pair deletion spanning from intron 5 to exon 7 (NM_014080.4:c.513 + 53_818del, expected to be deleterious). No other variants were found by exome sequencing.
Patient 2 had the p.M866R and p.F966SfsX29 variants, both known to cause complete loss of activity (2), and otherwise unrevealing targeted exome sequencing results. Unexpectedly, patient 3, although euthyroid, had the same DUOX2 mutations as his sister. Patient 4 harbored two previously reported variants, including p.F966SfsX29 and a p.P303R substitution; his exome data did not reveal any other relevant variants. Patient 5 is homozygous for the p.P303R variant. Patient 6 harbors the same mutations as her son (patient 4).
Discussion
To the authors' knowledge, patient 1 is the first infant with DUOX2 deficiency presenting with a life-threatening compressive goiter. The respiratory distress disappeared within three days of LT4 substitution, confirming the exquisite sensitivity to replacement therapy of rapidly growing hypothyroid goiters in fetuses and infants (4), which could be explained by the lack of fibrosis in goiters of rapid onset. Consequently, because of the response to LT4 therapy, decompressive surgery was unnecessary.
Patients 2 and 3 are siblings with the same DUOX2 mutations, yet they have completely discordant phenotypes, demonstrating that ethnicity alone does not always account for clinical heterogeneity (2).
Patient 4 has persistent mild congenital hypothyroidism, typical of DUOX2 deficiency. Surprisingly, the mother bears the same DUOX2 variants as her son, yet she is euthyroid. In this family, the finding of the p.P303R variant, which reduces enzyme activity by only 25%, is consistent with its high prevalence among patients with thyroid dyshormonogenesis (2). Although below the newborn screening cutoff, the TSH at 14 mIU/L of patient 5, who is homozygous for p.P303R, is consistent with the hypomorphic character of this variant. Importantly, careful serum TSH monitoring during eventual pregnancies is indicated in this patient.
Patients 3 and 6 are the first reported cases of bi-allelic pathogenic mutations of DUOX2 associated with lifelong euthyroidism. Thus, subjects with biallelic DUOX2 mutations differ by age at presentation (congenital or acquired), transient or permanent hypothyroidism, presence or absence of goiter (5), and, as shown here, the very presence of hypothyroidism. A transient phenotype could result from the compensation of peroxide generation by the DUOX1/DUOXA1 complex when thyroid hormone requirements fall with age. Such compensation may be epigenetically regulated: the locus on chromosome 15 that contains DUOX1 and 2 and their respective maturation factors is a site of DNA methylation (6), and demethylating agents increase DUOX1 and 2 expression in thyroid cells (7). Perinatal iodine overload reportedly masked congenital hypothyroidism at newborn screening in one of two siblings with biallelic DUOX2 mutations (8), but there was no such history in the discordant siblings reported here.
In summary, this search for DUOX2 deficiency in children from Quebec led to the discovery of a novel deletion and broadens the phenotypic spectrum of this condition, which spans from life-threatening compressive goiter in infancy to lifelong euthyroidism. Genetic and environmental modifiers accounting for this wide spectrum remain to be identified. Infants presenting with rapidly developing goiters with hypothyroidism due to dyshormonogenesis should first be treated with LT4 replacement, which can avoid decompressive surgery. Total thyroidectomy should never be considered in these patients because goiters due to DUOX2 deficiency can often resolve in response to LT4 substitution (9). Moreover, hypothyroidism associated with DUOXA2 mutations may also resolve without therapy (10). Hence, retesting the affected individuals at regular intervals is indicated.
Footnotes
Acknowledgments
Research in pediatric thyroid diseases at the Ste-Justine Hospital is supported by private donations to J.D. and G.V.V. (Girafonds, Ste-Justine Foundation). X.D.D. is supported by the “Fonds de la Recherche Scientifique” (FRS-FNRS) and the Doctor J.P. Naets Fund, managed by the King Baudouin Foundation (Belgium). The study was further supported by a European Society for Paediatric Endocrinology (ESPE) Research Unit Collaborative Project Grant to G.S. and J.D.. The costs of exome sequencing in patient 1 were covered by the Academic Fund of the Endocrinology Service of the Sainte-Justine Hospital. We thank all patients and their families for their cooperation. In addition, we thank Dr. Candice Hoste for sequencing DUOX2 in patients 2 and 3, Dr. Béatrice Gulbis for urinary iodine measurements, and Dr. Jean-Claude Décarie for help with interpretation of the MRI.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1