
Abstract
Background:
Graves' disease is associated with thyrotropin receptor (TSHR) antibodies of variable bioactivity. Recently, antibodies have been characterized that bind to the cleavage region of the TSHR ectodomain (C-TSHR-Ab), and their ability to induce thyroid cell apoptosis in vitro via excessive cell stress involving multiple organelles was demonstrated.
Methods:
To investigate the in vivo effects of C-TSHR-Ab, first a murine monoclonal antibody (mAb) directed against residues 337 to 356 of the TSHR cleavage region was developed, and then it was injected into mice.
Results:
These injections caused reduced serum total triiodothyronine and thyroxine and increased TSH levels compared to control mAb-injected mice. The C-TSHR-mAb induced histological evidence of endoplasmic reticulum stress, mitochondrial stress, and apoptosis in the thyroid glands. C-TSHR-mAb-mediated apoptosis was associated with cellular infiltrates consisting mostly of macrophages, dendritic cells, and neutrophils, while T- and B-lymphocytes were scarce. In addition, in the treated mouse thyroid tissue, hyper-citrullination of histone H3 was also found. This is known to occur via peptidylarginine deiminase 4 and plays an important role in the formation of neutrophil extracellular traps, which are likely to be partly responsible for thyroid infiltration, as seen in many autoimmune diseases. Examination of thyroid tissue from patients with Graves' disease also showed increased stress and some thyrocyte apoptosis compared to normal thyroid tissues.
Conclusions:
The fact that the C-TSHR-mAb induced accumulation of macrophages, neutrophils, and dendritic cells indicates that innate immunity plays a central role in shaping the adaptive immune response to the TSHR. In addition, this study provides further evidence that the hinge region of the TSHR ectodomain is intimately involved in the immune response in autoimmune thyroid disease.
Introduction
The thyrotropin receptor (TSHR) is a major autoantigen in autoimmune thyroid disease in both Graves' disease (GD) and Hashimoto's thyroiditis, demonstrating a thyroid inflammatory response through the action of thyroid specific T cells and multiple autoantibodies. Moreover, the immunologic processes involved, especially in GD, have one unique characteristic feature: the presence of autoantibodies to the TSHR, which have both linear and conformational epitopes (1). Three types of TSHR antibodies (stimulating, blocking, and neutral) with different functional capabilities have been described in such patients, which induce different signaling effects varying from thyroid cell proliferation to thyroid cell death (2 –6). The so-called neutral type of TSHR-Ab recognizes linear epitopes in the hinge region of the TSHR, including the “cleavage” region of the receptor ectodomain (amino acid residues 316–366). Although they are called neutral because they are unable to compete with TSH for binding to the TSHR and unable to stimulate Gαs and generate cyclic adenosine monophosphate (cAMP), it has been shown that these antibodies are capable of initiating a cascade of signaling events that lead to programmed cell death (1,3,6 –9).
The establishment of animal models of GD by immunization with TSHR antigen has confirmed its pathogenic role, and GD is therefore the result of a breakdown in TSHR tolerance (7,8). The frequency of neutral TSHR antibodies in GD ranges from 30% to 90%, based on linear epitope binding to known amino acid residues (3,4,7,8,10,11). Although the clinical importance of these antibodies has not been well studied, their pathophysiological role was characterized in an in vitro thyrocyte culture system (7,8). These data showed that neutral TSHR autoantibodies induced apoptosis through multiple stress signaling pathways (4 –7) and that the failure to sustain key signaling pathways led to thyrocyte death. It was hypothesized that neutral-mAbs would have similar effects in vivo, and therefore a mouse monoclonal antibody (mAb) recognizing a TSHR cleavage (hinge) epitope was developed for administration to mice. Here, the findings are described, which help delineate the potential pathogenic role of such antibodies, we found evidence of reactive oxygen species (ROS) generation and severe stress in the cellular organelles, causing thyroid cell death via apoptosis. Cellular infiltrations in the thyroid glands of treated mice included mainly innate immune cells such as macrophages, neutrophils, and NK cells. These observations suggest that cellular apoptosis may in turn orchestrate inflammatory autoimmune reactions in the thyroid.
Methods
Enzyme-linked immunosorbent assays
To detect mouse immunoglobulin G (IgG), a solid-phase indirect enzyme-linked immunosorbent assay (ELISA) was developed using two peptides (P#1 and P#2). Briefly, 96-well plates were coated with specific peptides (2 μg/mL) overnight, blocked with 5% bovine serum albumin (BSA), and incubated with diluted mouse serum samples in blocking buffer. A known mAb (Tab-16) was also run dose dependently in the same plate. Incubation with the horseradish peroxidase (HRP)-conjugated rabbit anti-mouse or anti-hamster antibody, washings, substrate reactions, and plate reading were performed according to a standard ELISA protocol. Further, to confirm antibody binding to the TSHR cleavage region, a peptide inhibition ELISA was also performed in the presence or absence of P#1. A reduction of mean optical density (OD) in the presence of the peptide compared to an irrelevant peptide indicated specific binding of IgG. Mean OD values ±3 standard deviations were used to determine a cutoff level to generate seropositivity.
Flow cytometry
To confirm the specific binding of purified mouse IgGs to the cleavage region further, serum samples from immunized mice were incubated with fixed and unfixed TSHR over expressed CHO cells (JPO9) by flow cytometry (3). Antibodies to cleavage region (linear epitope) recognized both fixed and unfixed cells, whereas isotype control as well as stimulating antibodies (MS1) was not.
Western immunoblot
To confirm the high specificity of the MC1 antibody binding to the TSHR cleavage region, Li-COR Western immunoblot assay was performed using cell lysates from FRTL5 thyrocytes and human thyroid follicular cells from stem cells (HTFS) (12).
ROS assay in live cells and tissues
In these assays, H2DCF-DA (total ROS [tROS]) or H2R123 (mitochondrial ROS [mROS]) oxidizes a nonfluorescent probe to a fluorescent probe that is detected by either flow cytometry or spectrofluorometry (3). Briefly, synchronized adherent cells were loaded with 50 μ
Luciferase reporter assay using CHO-TSHR cells
To measure Gsα activity, a CHO-TSHR reporter assay was used, as described recently (6). Gsα has been known to activate cAMP. Briefly, a fixed number of cells (3 × 104) on a 96-well plate were used to assess luciferase activity. In the presence of TSH and TSHR-stimulating or blocking antibodies, increasing concentrations of MC1-mAb were incubated, and then luciferase activity was measured.
Treatment of mice with mAbs
BALB/C/j female mice (n = 15; 6–8 weeks old) were given six injections of 100 μg of MC1 purified IgG over 12 days by intraperitoneal injection (Supplementary Fig. S1). Isotype-matched control mAb (IgG3 from a hybridoma recognizing KLH; BD Biosciences) was used for injecting control mice (n = 15; 6–8 weeks old). Two days after the last injections, blood was collected by cardiac puncture, and serum was prepared accordingly. Thyroid glands were harvested and frozen in OCT compound (Tissue-Tek; Sakura) and stored at −80°C for immunohistochemistry (IHC). These injections were performed in three groups of mice (n = 5) and in three independent experiments, each time with control mice (n = 5). Approval of these studies was provided by the Institutional Animal Care and Use Committee.
Serum triiodothyronine, thyroxine, and TSH assays
Serum samples from mice were analyzed for triiodothyronine (T3), thyroxine (T4), and TSH levels by a commercially available multiplex bead array (Milliplex Map Kit; Millipore) using Luminex xMAP instruments (Millipore).
IHC of rat thyrocytes and mouse thyroid tissues
IHC was performed according to protocols described earlier (6). Adherent FRTL5 thyrocytes were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde, permeabilized in 90% methanol, and blocked for two hours in blocking buffer. The cells were then incubated overnight at 4°C with specific antibodies diluted in blocking buffer. After washing, the slides were incubated with fluorescently conjugated secondary antibodies for one hour with appropriate dilution, washed three times in PBS, mounted with mounting medium containing nuclear dye (4′,6-diamidino-2-phenylindole [DAPI]), and visualized immediately under digital or confocal microscopy or both (6). Fresh frozen thyroid thin sections (5 μm) were blocked with 5% BSA in PBS and incubated at 4°C overnight with conjugated primary antibodies, washed in PBS, mounted as DAPI staining, sealed with clear nail polish, and observed under a microscope (6). In case of unconjugated primary antibodies, additional incubation was added with fluorescent-labeled secondary antibodies.
Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling assay
DNA cleavage in apoptotic cells can be detected in situ in fixed cells or tissue sections using the terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) method. TUNEL is highly selective for the detection of apoptotic cells but not necrotic cells. The TUNEL Andy Fluor™ 488 apoptosis detection kit from GeneCopoiea was used to detect apoptotic cells in frozen thyroid gland thin fixed sections according to the protocol provided with the kit.
Quantification of live fluorescence and IHC images
The recorded images were loaded into Adobe Photoshop (Adobe Systems) for analysis, in which different colors were visually quantified by two independent observers. The number of cells positive for both the target color and nuclear staining (DAPI) was considered positive for fluorescence and digitally recorded to prevent multiple counts. Relative fluorescence intensity (RFI) or unit was calculated, as described recently (6). The number of cells positive for distinct co-localized colors was also digitally recorded. CellProfiler and Image-Pro software were used to discriminate between different colors, including co-localization and average RFI measurements.
IHC of paraffin-embedded human thyroid tissues
Human thyroid tissues from GD patients (n = 3) and healthy individuals (n = 3) were collected in 10% neutral-buffered formalin. After fixation, a paraffin block was made using the standard protocol. IHC was performed using a vector IHC kit (Vector Laboratories Ltd.). Briefly, paraffin-embedded human thyroid tissues were deparaffinized, rehydrated, antigen unmasked (retrieved), blocked with blocking buffer, incubated with primary antibodies, and incubated with the appropriate HRP-conjugated secondary antibodies. After washing, DAB substrate solution was applied for color development, counterstained with hematoxylin, and finally mounted by cover slip with Permount mounting solution.
Antibodies used in IHC experiments
Antibodies to Caspase 3A, PD1, BiP, CHOP, and Grp94 were from Cell Signaling Technology. HSP 90, HSP 60, and 8-OHdG (DNA-damage proteins) were from StressMark. Peroxiredoxin-3 (Per-3) and BAX were from Aviva Biosciences. Ly6G, PAD4, F4/80, CD11c, CD4, CD8, FoxP3, CD25, B220, CD3, CD16, CD69, and isotype control-mAbs (IgG3) were all from BD Biosciences. K1-70 TSHR-blocking mAb and citrullinated protein histone 3 (Citr-H3) antibody were from RSR Ltd. and Abcam, respectively.
Statistical analysis
Two-tailed paired t-tests were used to evaluate the statistical significance of differences in means for continuous variables. p-Values for fold changes and percentage increases were calculated after log2 transformation. A p-value of ≤0.05 was used to determine statistical significance. Data are means ± standard error of the mean.
Results
Development of a mAb to the TSHR cleavage region (MC1)
To demonstrate immunogenicity of linear TSHR cleavage epitopes, first Balb/c/j mice were immunized with two peptides (amino acids 322–341 and 337–356). The immunogenic peptide sequences were chosen using EMBOSS Explorer antigenicity software. Mice (n = 5) were immunized with 100 μg KLH-conjugated peptides in CFA (13). After the last immunizations, sera were collected from mice and analyzed for antibodies by peptide ELISA. Out of five mice sera, one showed extra-strong binding with amino acids 322–341. Peptide (337–356) did not initiate a good immune response. The mouse with the highest titer by peptide ELISA was selected for hybridoma cell production (Supplementary Fig. S2A). Purified IgG from the culture supernatant demonstrated strong specific peptide binding, which was further confirmed by fluorescence-activated cell sorting analysis (Supplementary Fig. S2B), indirect immunofluorescence staining (Supplementary Fig. S2C) and Western blot analysis of FRTL5 thyrocyte lysates, and HTFS (Supplementary Fig. S2D). Furthermore, peptide inhibition ELISA (Supplementary Fig. S2E) demonstrated the high specificity of the mouse IgG binding to the peptide. Determination of IgG isotyping showed MC1 to be IgG3 (not illustrated).
Functional characterization of neutral mAb MC1 in vitro
As expected from previous studies (3), MC1 induced both tROS and mROS in FRTL5 thyrocytes (Supplementary Fig. S3A and B) in a dose- and time-dependent manner (Supplementary Fig. S3C). By contrast, a control isotype mAb (cont-mAb) did not induce excessive ROS. It was previously shown that excessive ROS production in cells can induce cell death and DAPI staining disclosed nuclear condensation and nuclear breakdown similar to apoptotic bodies, which indicated thyroid cell death by apoptosis after exposure to MC1 (Supplementary Fig. S3D). This was confirmed with detection of the caspase 3a catalytic unit, a selective marker of apoptosis, which demonstrated cytosolic expression of the protein along with mROS (Supplementary Fig. S3D). Specificity of different types of ROS was confirmed by a non-selective antioxidant, MnTBAP, which suppressed both tROS and mROS, and a selective inhibitor of mROS, SkQ1, was used (Supplementary Fig. S4). MC1 did not influence the function of TSH, TSHR-stimulating, and -blocking antibodies, as shown by the luciferase reporter assay in CHO-TSHR overexpressed cells (Supplementary Fig. S5).
MC1 lowers serum T3 and T4 and increases TSH in mice
To demonstrate the biological functional effect of MC1 binding to TSHR in vivo, MC1-mAb was injected into two groups of BALB/C mice, as described in Figure 1. Serum T3, T4, and TSH were measured in both MC1- and control-mAb-injected mice. Serum T3 and T4 (Fig. 1A and B) levels were significantly reduced in MC1-treated mice compared to the control group, while serum TSH was increased (Fig. 1C). Out of 15 mice, 13 (87%) showed lower levels of T3 and T4, whereas 12/15 (80%) showed higher level of TSH in sera. None of the control-mAb-injected mice showed reduced T3 and T4. Only one mouse had a higher TSH level. These data indicate that MC1 induced a state of mild hypothyroidism.
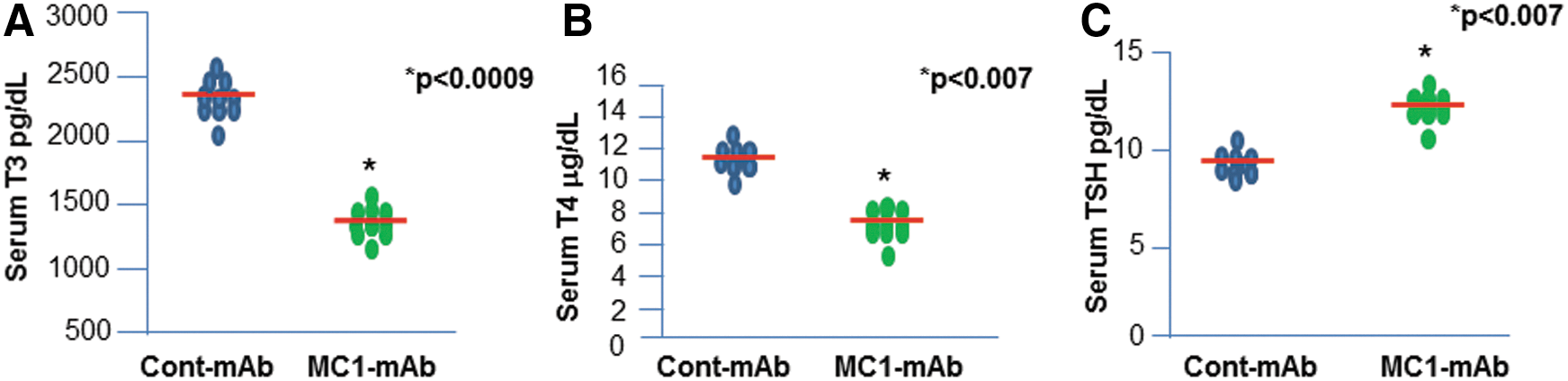
MC1 lowers serum triiodothyronine (T3) and thyroxine (T4) and increases thyrotropin (TSH) in mice. MC1-treated mice show reduced serum T3 (p < 0.0009) and T4 levels (p < 0.007) compared to control monoclonal antibody (mAb)-treated mice, as shown in (
MC1 induces thyrocyte apoptosis in mouse thyroid glands
MC1 was detected in the thyroid glands of MC1 injected mice by fluorescence staining with an antibody (Fig. 2A). The presence of MC1 indicated that the MC1 antibody can target thyrocytes. Quantitative image analyses also indicated accumulation of significant levels of MC1 antibody in the thyroid glands (Fig. 2D). Assuming the induction of hypothyroidism was secondary to thyrocyte death, the TUNEL assay was examined for apoptosis in thyroid tissues from MC1-treated mice, and positive staining was found for apoptotic cell death in thyrocytes (Fig. 2B). Not all thyrocytes in different follicles were apoptotic, and there was a heterogeneous distribution of apoptotic cells. Quantitative image analyses revealed that the staining intensity was significantly higher in MC1-treated mice than in controls, which showed almost no staining (Fig. 2E). Out of 15 mice, 13 (87%) showed positive TUNEL staining, whereas none of the mice in the control group showed any positive staining. To confirm that apoptosis was the main culprit for orchestrating cell death in the thyroid glands, the caspase 3A (active) catalytic unit in the thyroid tissues was examined (Fig. 2C); quantitative analyses of thyrocytes indicate that MC1 induced caspase 3A activity to a significantly higher extent compared to the levels found in control mice (Fig. 2F). Caspase 3A staining was also scattered throughout follicles in the thyroid tissue.
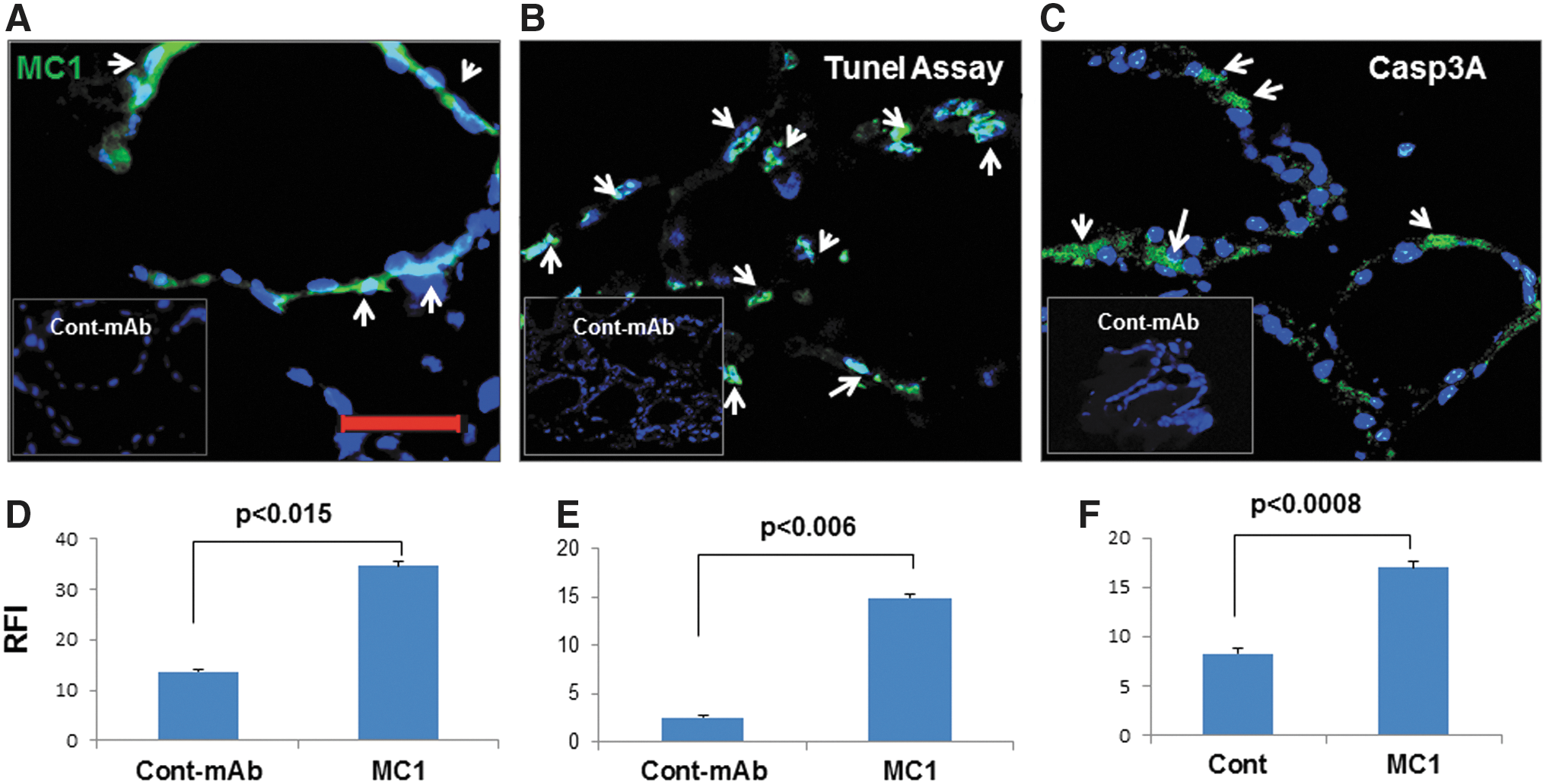
MC1 induces thyrocyte apoptosis in mouse thyroid glands. The accumulation of MC1 antibody in thyroid follicles after injection of the MC1 antibody is shown in (
MC1 induces ROS and endoplasmic reticulum stress
It is well known that cAMP is the key to thyrocyte homeostasis and that neutral TSHR antibodies such as MC1 are unable to induce cAMP generation via the activation of G proteins. Therefore, it was suspected that MC1 would also induce ROS in thyroid tissues in vivo. Indeed, activation of ROS was markedly increased in MC1-treated mice (Fig. 3A and B). Furthermore, ROS is known to be a potent stressor for both the endoplasmic reticulum (ER) and mitochondria and can induce protein misfolding responses (14). As a result of ROS induction by MC1, the ER stress markers PD1, BiP, and HSP90 were found to be induced in treated thyroid tissues compared to control tissues (Fig. 3A and B). Consistent with the observed ROS induction, positive TUNEL staining was observed in the treated thyroid glands—an observation that was absent in controls.
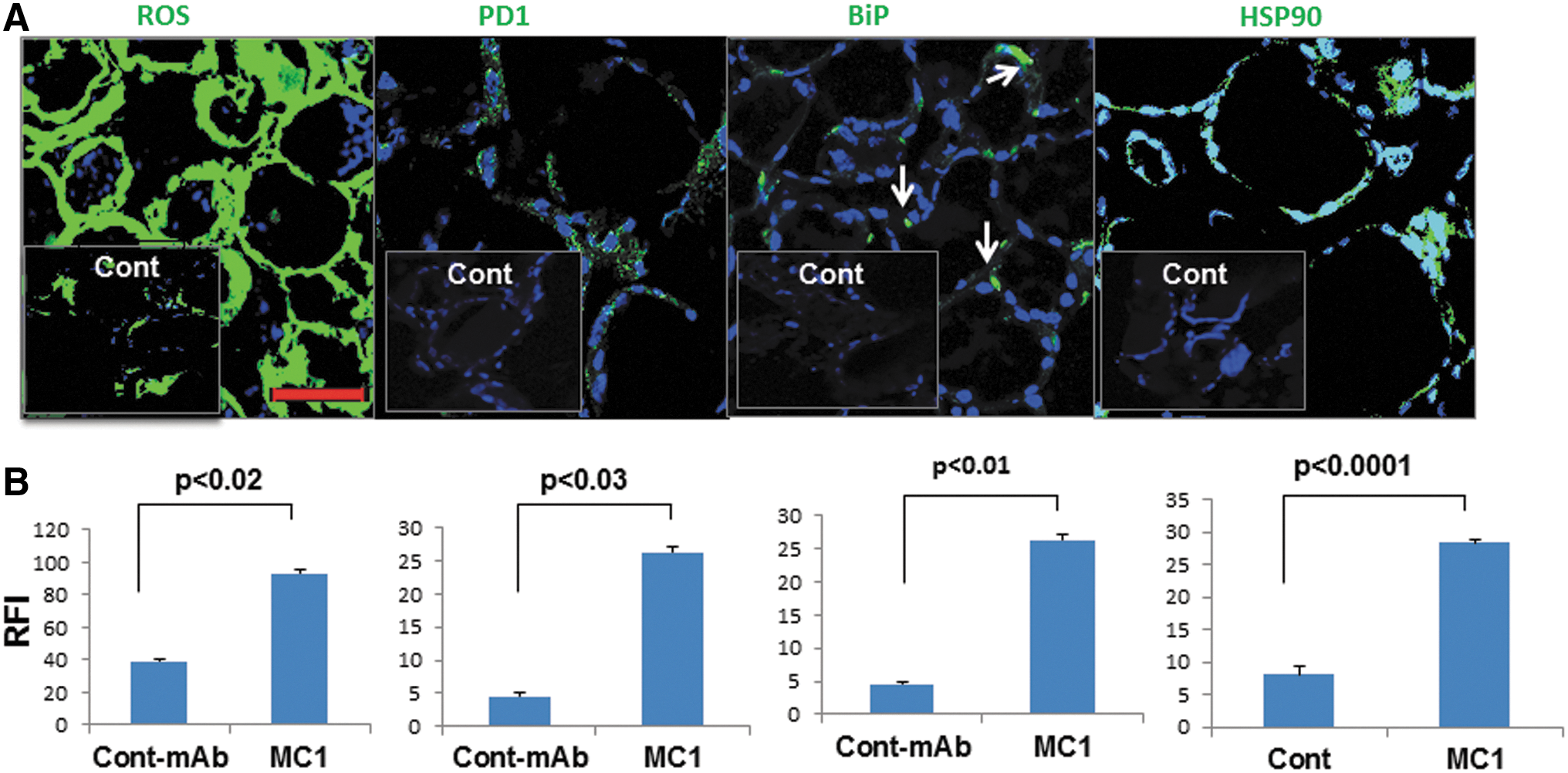
MC1 induces reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress. Expression of ROS, PD1, BiP, and HSP90 is shown in the upper panel of (
MC1 induces mitochondrial stress/DNA damage but decreases Per-3 in thyroid glands
HSP60 is a selective marker of mitochondrial stress, and Per-3 is an essential antioxidant protein that neutralizes excessive ROS in the mitochondria (15). Excessive ROS is particularly involved in thyroid cell DNA damage because they produce more H2O2 than other cells (16,17). While immunostaining of the HSP60 and DNA damage marker, 8-OXDG was significantly increased in thyroid glands from MC1-treated mice (Fig. 4A and B), Per-3 failed to show any increase in its expression. In fact, it appeared to show the opposite effect; quantitative image analyses revealed that MC1 treatments reduced the expression of Per-3 significantly compared to control mice (Fig. 4A and B). Mice with TUNEL-positive thyroid glands were mostly positive for mitochondrial stress and DNA damage marker and negative for Per-3, whereas the opposite was found in control-mAb-injected mice.
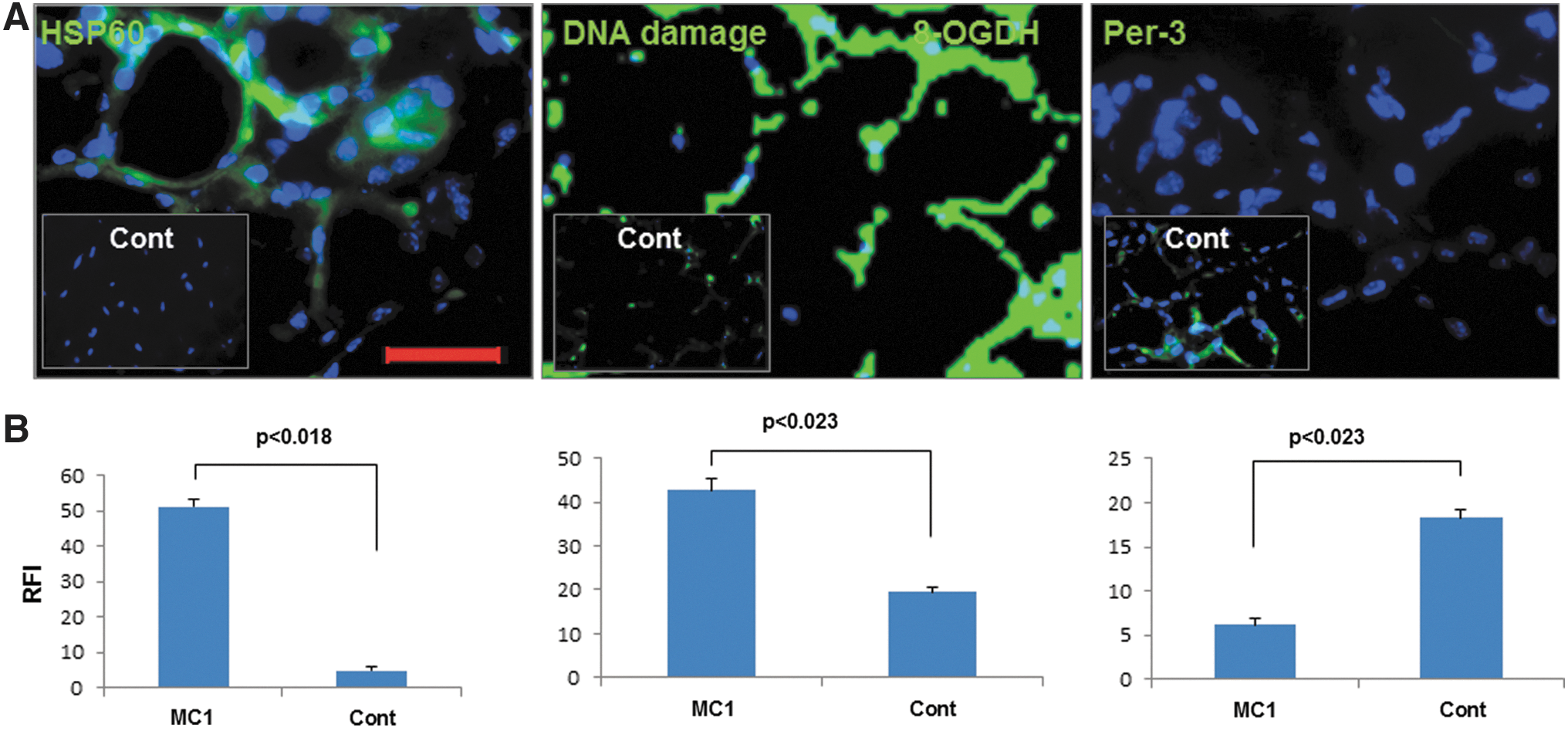
MC1 induces mitochondrial stress/DNA damage but decreases peroxiredoxin-3 (Per-3) in thyroid glands. Expression of HSP60, DNA damage protein, and Per-3 is shown in the upper panel of (
MC1 induces neutrophil infiltrates in thyroid glands
Apoptotic events in epithelial cells are early initiators of inflammation (18). The released products by apoptotic cells invite innate immune cells to scavenge harmful apoptotic debris (19). The presence of apoptotic thyrocytes in the thyroid follicles of MC1-treated mice would be expected to induce infiltrates of innate immune cells such as neutrophils, macrophages, dendritic cells, and many other types of lymphocytes. Using specific fluorescence-labeled mAbs, some of these inflammatory cells were characterized in the thyroid glands of the treated mice. Expression of Ly6G (neutrophil) was mostly observed in the cellular infiltrates of the inter-follicular spaces (Fig. 5A and D). Protein arginine deiminase (PAD) type 4 is believed to be a marker for neutrophil extracellular trap (NET) or NEtosis (i.e., extracellular DNA release), which was also present (20) (Fig. 5B and E). Citr-H3 is an immunogenic protein and can be a target for infiltrates (21). Significantly elevated staining of Citr-H3 protein (Fig. 5C and F) was also observed in the inter-follicular spaces of thyroid tissues of MC1-treated mice compared to controls. Staining for Ly6G, PAD4, and Citr-H3 were positive in 13/15 (87%) MC1-injected mice, whereas none of the control mice showed any staining.
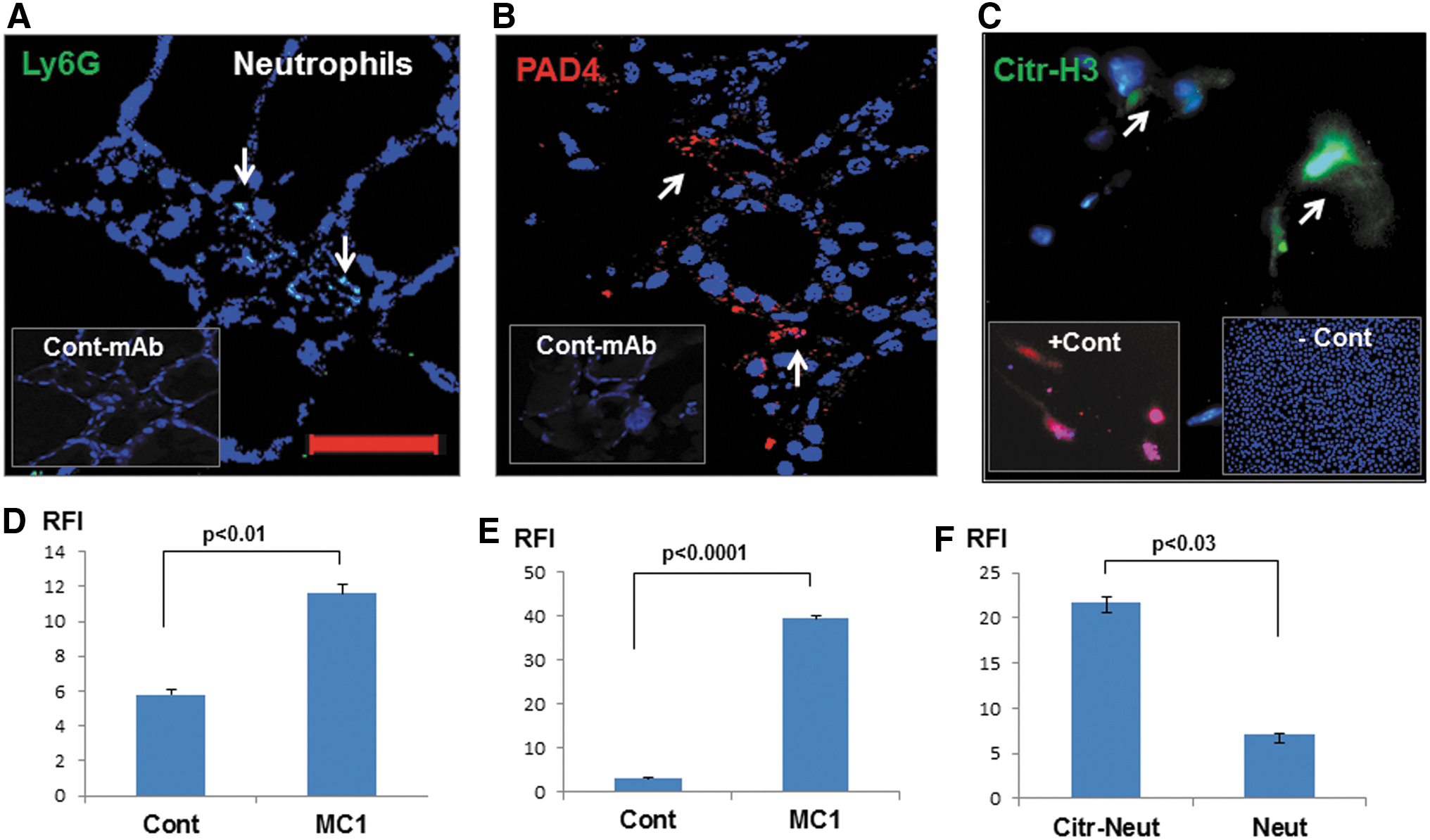
MC1 induces neutrophil infiltrates in thyroid glands. Neutrophil infiltration (Ly6G) was found to be increased in thyroid tissues (
MC1 induces macrophages, dendritic cells, and other lymphocyte infiltrates in thyroid glands
Induction of macrophage (F4/80 and CD14) infiltrations in the inter-follicular spaces was also evident in the MC1-treated mouse thyroid (Fig. 6A and B). Interestingly, macrophages were also seen in contact with thyrocytes and in the thyroid colloid (Fig. 6A, white arrows). Such accumulation of macrophages was also observed in apoptotic tissue debris. Dendritic cells (CD11c) were observed in the infiltrates, and although they were significantly increased in MC1-treated mice compared to controls (Fig. 6C and D), they were lower compared to macrophages. As for an adaptive immune response, T-lymphocytes (CD4+ and CD8+; Fig. 6E and F), Treg cells (CD25+ and FoxP3; Fig. 6G and H), B-lymphocytes (CD220; Fig. 6I and J), natural killer T cells (NK-T cells, CD16+CD3), and NK cells (CD16; Fig. 6K and L) we also analyzed in the thyroid tissues of MC1-treated mice, and some T and B lymphocytes were found in the infiltrates, but they were scarce and were in peripolesis (the process in which a cell attaches itself to another cell) and not observed in all inter-follicular spaces (Fig. 6E and F), which were occupied by macrophages. However, compared to control mice, both CD4+ and CD8+ lymphocytes were significantly increased in MC1-treated mice (Fig. 6F) along with infrequent B lymphocytes (Fig. 6I and J). NK-T cells (Fig. 6K) and NK cells (Fig. 6L) were also observed, but they were lower in numbers and infrequent in inter-follicular spaces. Both NK cells and NK-T cells were absent in control thyroid tissues (Fig. 6K, inset). Interestingly, scarce Th17 and activated T lymphocytes (CD69) were also observed only in the MC1-injected mice (Fig. 6M and N). All mice with TUNEL-positive thyrocytes were positive for cell infiltrates, and such findings were absent in the controls. Hematoxylin and eosin staining revealed damaged follicles (Fig. 6O, red arrows) and cell infiltrates in line with the immunostaining (Fig. 6O). Macrophages were also found that were mostly in the inter-follicular spaces and colloid of the thyroid follicles (Fig. 6O, black arrows).
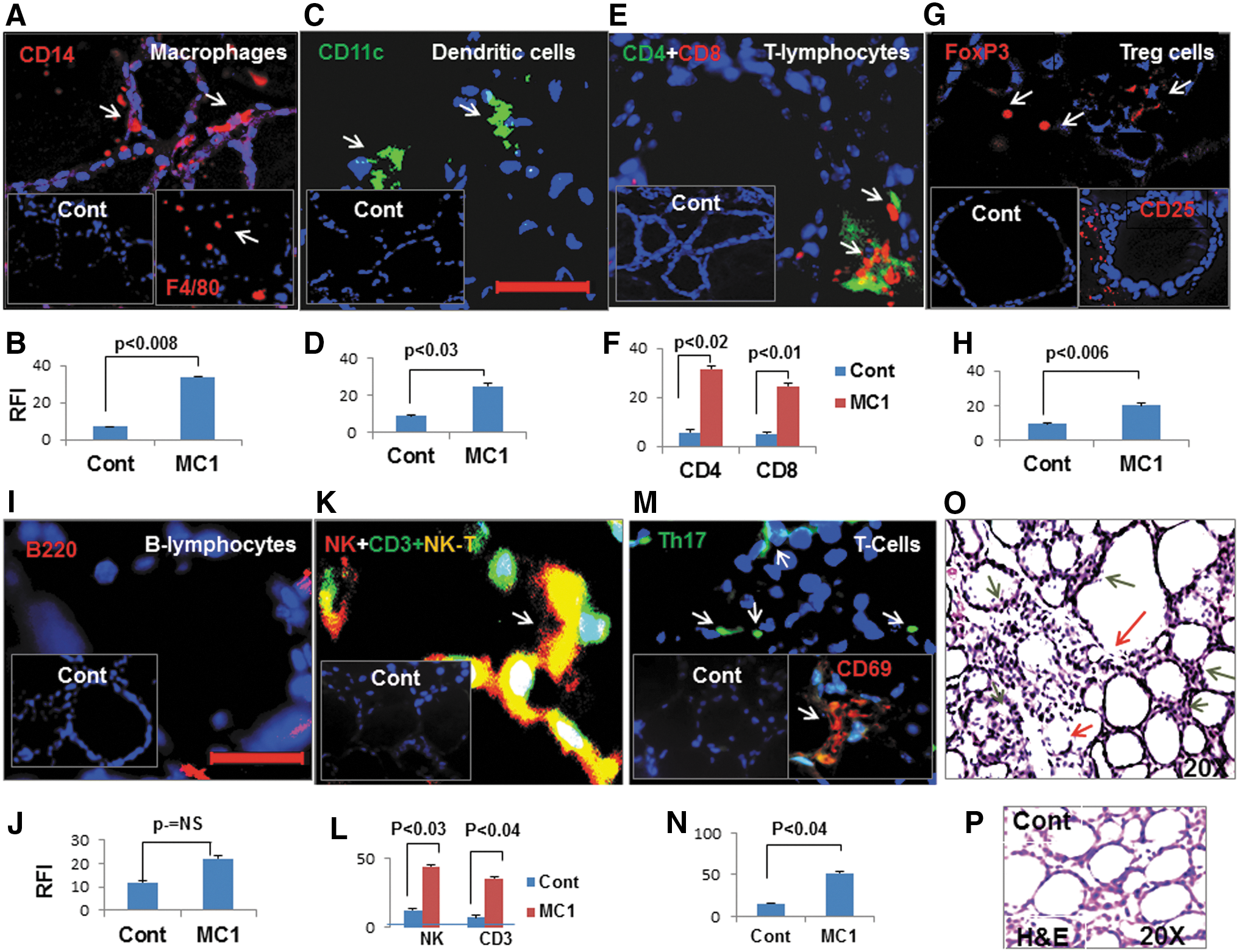
MC1 induces macrophages, dendritic cells, and other lymphocyte infiltrates in thyroid glands. CD14+ (red) macrophages (red, inset F4/80), CD11c+ dendritic cells (green), CD4+(green) plus CD8 T (red) lymphocytes, and FoxP3 Treg (red) or CD25+ T (red, inset) cells are shown in (
GD thyroid tissue shows increased ER stress, DNA damage, and apoptosis
Since patients with GD are known to generate TSHR antibodies, including “neutral” TSHR antibodies, GD thyroid tissues were analyzed to see if they reflected the histopathological observations found in the MC1-treated mouse model. Thyroid tissues were stained for markers of ER stress, DNA damage, and apoptotic proteins. Using specific antibodies to CHOP and Grp94 proteins, markers for ER stress proteins, significantly increased expression of these proteins was observed in the GD thyroid tissues (n = 3) compared to thyroid tissue from healthy individuals (n = 3; Fig. 7). Similarly, 8-hydroxyguanosine, a marker for DNA damage protein, was highly expressed in the GD thyroid tissues, and BAX, an apoptotic protein, was also found to be increased (Fig. 7).
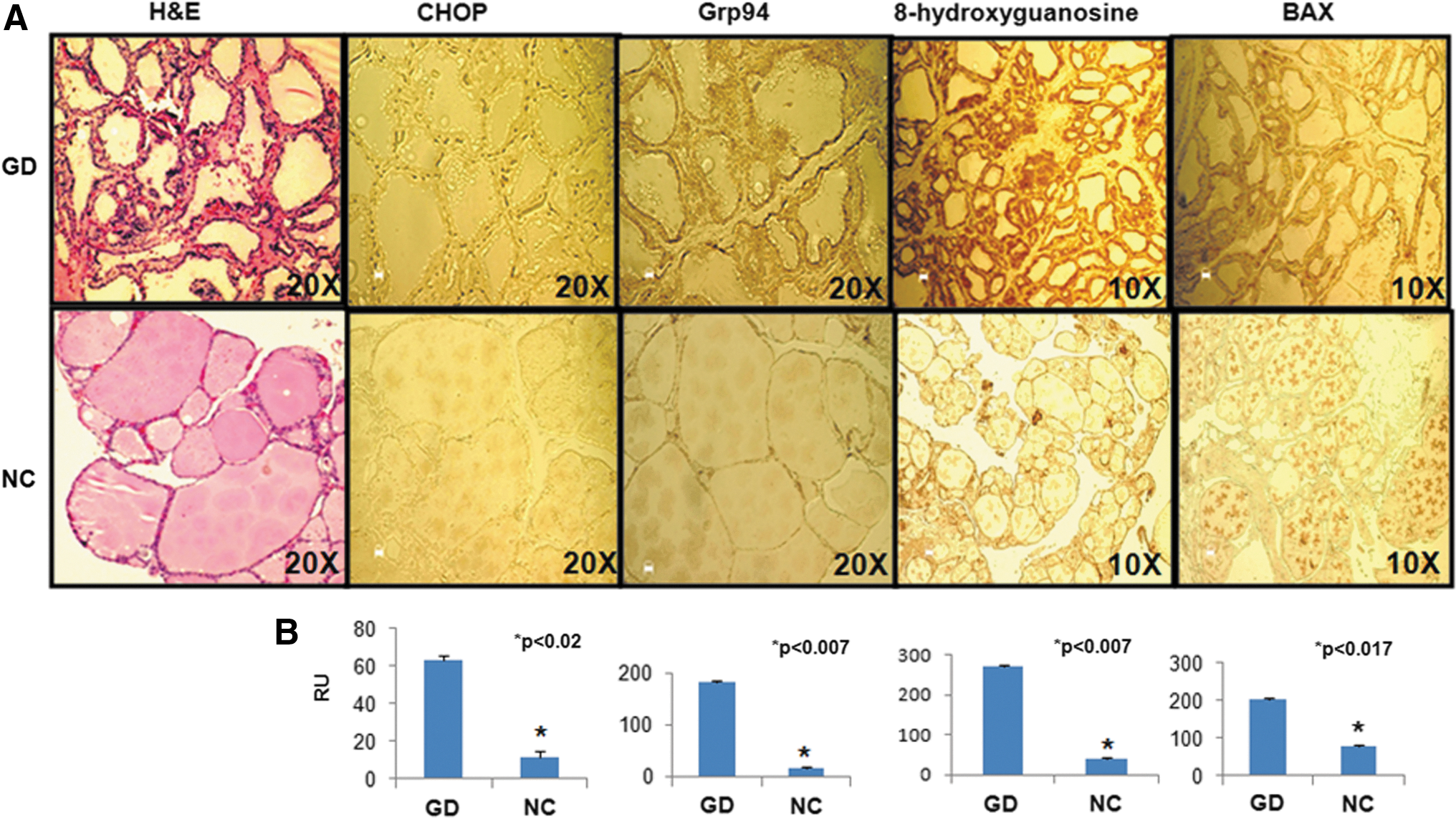
Graves' disease (GD) thyroid tissue shows increased endoplasmic reticulum (ER) stress, DNA damage, and apoptosis. H&E staining of inflamed thyroid tissue from GD (n = 3) is shown on the left in (
Discussion
Characterization of mAb interaction with the TSHR cleavage region in vivo confirmed earlier observations that this region may be an important immunogenic sequence, resulting in thyroid gland stress with apoptosis and a fall in thyroid hormone levels (3). The role of antibodies in the pathogenesis of GD is well known, and the use of well-characterized mAbs as tools for understanding the disease pathways in GD pathogenesis has allowed detailed structural analysis of the TSHR, which is the major autoantigen. A new murine mAb (MC1) was developed and selected for a specific cleavage region epitope, and it was found that it was able to induce mild thyroiditis, as evidenced by changes in thyroid function and thyroid cell survival. It has previously been shown that C-TSHR-mAbs are capable of inducing apoptosis in thyrocytes in vitro (3,5,8). Hence, these in vivo results show similar results using a new mAb that induced thyrocyte apoptosis. The mechanisms by which such TSHR-mAbs induce thyrocyte death have also been described in detail (6), which led to the hypothesis that similar mechanisms may be involved in the current in vivo model.
Disturbances in the normal redox state of cells can cause toxic effects through the production of peroxides and free radicals or ROS that damage all components of the cell, including proteins, lipids, and DNA. Since antioxidants can prevent thyrocyte death via scavenging ROS, it was possible to pinpoint ROS as the main culprit responsible for the initiation of cell death (3,5,6). These findings were further supported by the observations that ER and mitochondrial stress played a role in this cell-death process (3,5). ROS generated from mitochondria can induce both mitochondrial and ER stress in vitro (3,5). Similar findings were also observed in vivo with C-TSHR-mAb in mice. Interestingly, mitochondrial stress responses prevailed over ER stress in vivo, as mitochondrial ROS can induce mitochondrial DNA damage and can also reduce mitochondrial defense mechanisms (22). In fact, the observations of increased expression of HSP60, DNA damage protein, and reduced expression of Per-3, as a mitochondrial anti-defense protein, confirmed this interpretation. Clearly, the C-TSHR-mAb was able to induce ROS and multicellular stresses, which then resulted in thyroid cell death, since oxidative stress from oxidative metabolism causes DNA base damage as well as strand breaks (23). The increased DNA damage protein in thyrocyte cytosol from thyroid glands of mice treated with C-TSHR-mAb indicates that mitochondrial ROS induced mitochondrial DNA (mtDNA) damage. Mitochondrial DNA is located in close proximity to the respiratory chain, which is the main cellular source of ROS, and oxidative stress can lead to the degradation of mtDNA (23), thus compromising mitochondrial functions and activating mitochondrial apoptotic proteins, which then orchestrate cell death (24).
When cells die in vivo, they may trigger an inflammatory response. This response can itself add to the tissue damage, and this phenomenon may contribute to the pathogenesis of a number of diseases (18). Injured cells release “danger signals” that alert the host to cell death. These signals are recognized by certain receptors of host cells that stimulate the generation of proinflammatory mediators. The resulting mediators then orchestrate the inflammatory response inviting cellular infiltrates (18). An example of antibodies inducing such reactions comes from the examination of lupus IgG that induce neutrophil infiltration in multiple tissues and then cause tissue damage in a lupus mouse model (25). In addition, it is well known that macrophages play an important role in the clearance of dying and dead cells (26). Both neutrophil and macrophage infiltrations were also observed in the thyroid tissues of the treated mice. The presence of NET formation was also confirmed at the tissue level. These observations suggest that C-TSHR-mAb (MC1) induces neutrophil and macrophage infiltrates as part of an acute immune response. Further studies should aim to identify whether neutrophils or macrophages are the key players in this infiltration.
Tissue staining for cell subsets, which included dendritic cells, B lymphocytes, T lymphocytes, natural killer (NK) cells, NK-T cells, and Th17 cells, showed low numbers at the time point examined (two weeks), most likely because it was too early for a chronic inflammatory response. Their scanty presence indicates that two weeks of treatment with MC1 antibody was not sufficient to induce the changes that were observed in Graves' thyroid tissue. It is likely that antibodies to multiple TSHR epitopes may be responsible for the induction of chronic inflammatory changes, as seen in a mouse model of rheumatoid arthritis (27). However, these findings do highlight the possibility that the co-existence of multiple antibodies in Graves' patients may have a marked influence on the clinical phenotype. The findings of increased expression of ER stress, DNA damage, and apoptotic protein markers in GD thyroid tissue also indicate that they display signs of severe cellular stress as part of their chronic inflammatory changes. This would help explain the high number of patients with GD who develop hypothyroidism over time (28). Whether these stresses are the product of inflammatory changes or the action of multiple antibodies with different epitopes is unclear. Antibodies, as a constituent of immune complexes, may contribute significantly to triggering inflammation in a number of autoimmune diseases (29). Thus, the initial triggering event in the antibody transfer model could be the formation and deposition of TSHR–IgG immune complexes in the thyroid glands and inter-follicular spaces. Epitope specificity, affinity, and isotype of the accumulated antibodies on the TSHR and the presence of inducers of pro-inflammatory cytokines might contribute to the ultimate pathogenicity of the antibodies.
Conclusions
This study delineates the pathogenic role of cleavage region-specific TSHR antibodies, which induced thyroid cell ROS and severe stress in the cellular organelles, leading to thyrocyte death via apoptosis. The cellular infiltrations in the thyroid glands of C-TSHR-mAb-treated mice included mainly innate immune cells such as macrophages, neutrophils, and NK cells. These observations suggest that cellular apoptosis may orchestrate inflammatory autoimmune reactions in the thyroid glands of patients with GD.
Footnotes
Acknowledgments
This work was supported in part by DK069713 from the National Institutes of Health, the Segal Family Endowment, and the VA Merit Review Program (to T.F.D.)
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Fig. S1
Supplementary Fig. S2
Supplementary Fig. S3
Supplementary Fig. S4
Supplementary Fig. S5