
Abstract
Background:
Our genome sequencing analysis revealed a frameshift mutation in the shelterin gene TINF2 in a large family with individuals affected with papillary thyroid carcinoma (PTC) and melanoma. Here, we further characterized the mutation and screened for coding variants in the 6 shelterin genes in 24 families.
Methods:
Sanger sequencing was performed to screen for the TINF2 mutation in the key family. Quantitative reverse transcription-polymerase chain reaction (PCR) was used for TINF2 gene expression analysis. Exogenous expression and co-immunoprecipitation techniques were used for assessing TINF2 binding to TERF1. Relative telomere length (RTL) was quantified in DNAs from lymphocytes by using quantitative real-time PCR. Whole exome sequencing (WES) was performed in seven families with individuals affected with PTC and other cancer types. Screening for DNA variants in shelterin genes was performed by using whole genome sequencing data from 17 families and WES data from 7 further families.
Results:
The TINF2 mutation (TINF2
Conclusions:
The rare frameshift mutation in the TINF2 gene and the associated longer telomere length suggest that dysregulated telomeres could be a mechanism predisposing to PTC and melanoma. DNA coding variants in shelterin genes are rare. Further studies are required to evaluate the roles of variants in shelterin genes in thyroid cancer and melanoma.
Introduction
Nonmedullary thyroid cancer (NMTC) comprises thyroid cancers of follicular cell origin and accounts for >95% of all thyroid cancer cases. Papillary thyroid carcinoma (PTC) is the main form of NMTC, accounting for ∼75% to 80% of all thyroid cancers. It is estimated that 52,070 individuals in the United States will be diagnosed with thyroid cancer in 2019 (1). Although PTC is mostly sporadic, ∼5% is familial (2). A strong inherited genetic predisposition is suggested by case-control studies showing a three- to eight-fold increase in risk in first-degree relatives, this being one of the strongest examples of heritability in cancer (3,4). Over the past years, linkage and association studies have pinpointed several potential regions as harboring predisposing genes in NMTC families, including 1q21, 2q21, 4q32, 6q22, 8p23, 8q24, and 19p13.2 (5 –9).
Although a few candidate predisposing genes have been proposed, decisive evidence implicating specific genes has not been straightforward. Moreover, genome-wide association studies have pinpointed at least 10 loci having odds ratios of ∼1.2 to ∼1.8, indicating that low-penetrance predisposition variants play important roles (10). We tentatively propose that the genetic predisposition to NMTC is heterogeneous and perhaps predominantly caused by low-penetrance genes. However, high penetrance unique or very rare gene mutations may also play important roles in NMTC predisposition (7).
In our recent study analyzing 17 families by whole genome sequencing (WGS), we identified a frameshift mutation in the TERF1-interacting nuclear factor 2 (TINF2, also known as TIN2) gene in a large three-generation family with PTC and melanoma (11). The TINF2 gene encodes the TINF2 protein, which is one of the six known subunits of shelterin. They protect telomeres by allowing the cell to distinguish between telomeres and regions of DNA damage (12 –14). The other shelterin genes are: telomeric repeat binding factor 1 (TERF1, also known as TRF1), telomeric repeat binding factor 2 (TERF2, also known as TRF2), adrenocortical dysplasia protein homolog (ACD, also known as TPP1), telomeric repeat binding factor 2 interacting protein (TERF2IP, also known as RAP1), and protection of telomeres 1 (POT1).
The shelterin proteins TERF1, TERF2, and POT1 directly bind to telomeric DNA and are interconnected by TINF2, ACD, and TERF2IP. Although TINF2 does not directly bind to telomeric DNA, it is a critical part of shelterin through its interactions with TERF1, TERF2, and ACD. The structure of the shelterin complex and its functions in telomere maintenance has been extensively studied (13 –15). Mutations in the shelterin genes have been shown to have a strong impact on diseases related to cellular lifespan, particularly cancer (16 –18).
In this study, we characterized the truncating mutation TINF2
Methods
The study was approved by the Institutional Review Board at the Ohio State University (OSU), and all subjects gave written informed consent before participation.
Patients and controls
The key family in this study has been previously described (11,19). There were eight individuals affected with PTC; two of them had both PTC and melanoma. Among the remaining family members, two had melanoma only and two had chronic lymphocytic leukemia (CLL). An additional 10 individuals had benign thyroid disease (nodules or goiter), including 1 individual with goiter who also had both cutaneous and ocular melanoma, as well as breast cancer (Fig. 1 and Table 1).
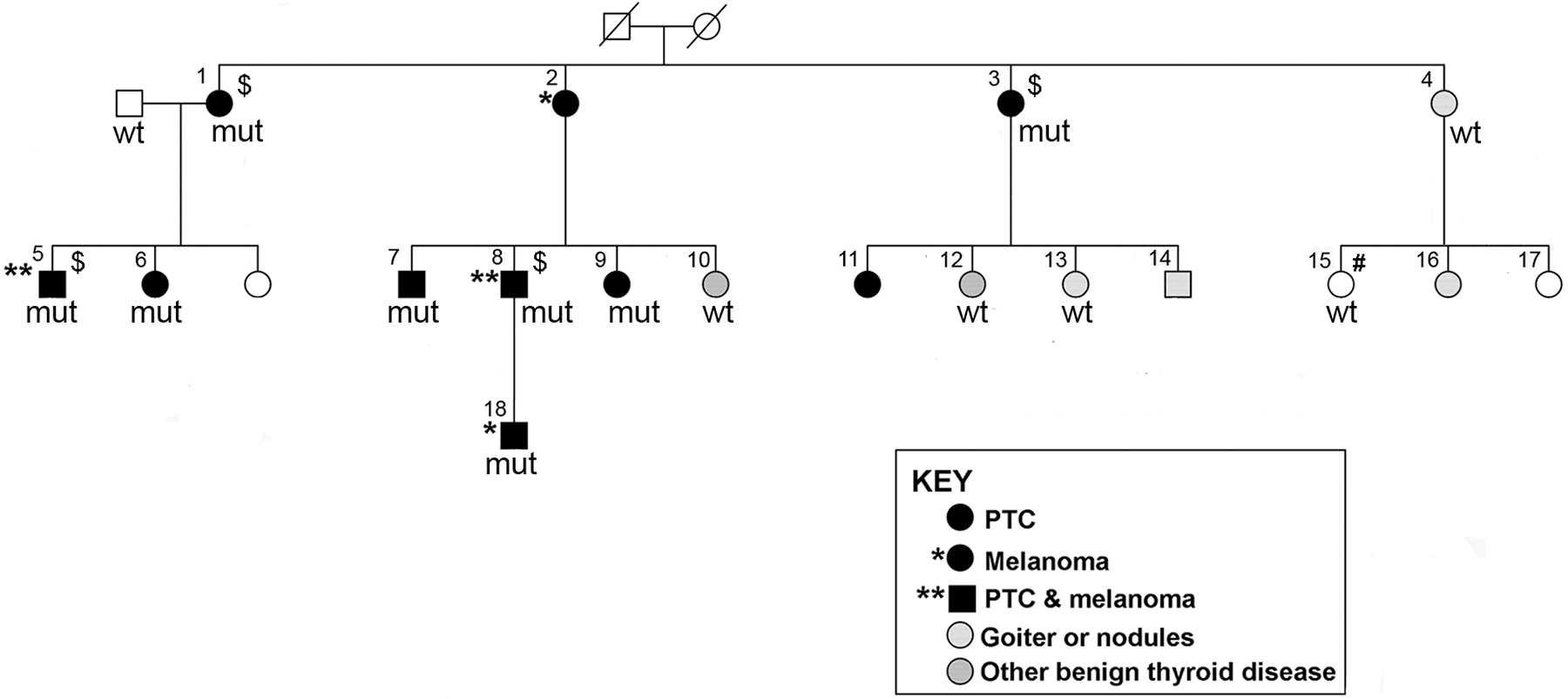
Pedigree of a family with a rare frameshift mutation in the TINF2 gene (in individuals with PTC and melanoma). Males are indicated by squares and females by circles. Individual IDs are labeled by using numbers. Deceased individual is displayed as a symbol with a diagonal line. mut, individuals with the TINF2 mutation (C/−, chr14: 24,710,239 on assembly GRCh37.p13,
Clinical Characteristics of the Available Individuals from the Family Studied
Ctr, control, by marriage; PTC, papillary thyroid carcinoma.
An additional seven families with at least three confirmed cases of PTC in close relatives were recruited. Affected individuals with PTC, melanoma, and other types of malignancy are labeled as indicated in Supplementary Figure S1. Cancer diagnoses were confirmed by pathological specimen review, medical records, detailed interview, or self-reported. Genomic DNA extracted from whole blood samples from affected individuals (n = 14, two per family) was used for whole exome sequencing (WES). Controls (n = 62) were individuals without clinically diagnosed thyroid cancer from central Ohio. All controls were of European descent (25 men, 37 women; median age 61 years, range 50 to 85 years). The control samples were chosen from an ongoing collection of control individuals recruited at OSU.
Genomic DNA extraction, polymerase chain reaction, and Sanger sequencing
Genomic DNA was extracted from whole blood samples according to standard phenol
Lymphoblastoid culture from whole blood
Blood lymphocytes were purified by using Ficoll–Paque PLUS medium (GE Healthcare) and density gradient centrifugation according to the manufacturer's protocol. Briefly, 10 mL of whole blood was diluted with 25 mL of RPMI-1640 growth medium (GIBCO) and layered onto 10 to 14 mL of Ficoll–Paque PLUS medium in a 50-mL tube. The sample was centrifuged at room temperature for 30 minutes, and the layer of lymphocytes was transferred to a new tube and washed with RPMI-1640 medium. The cell pellet was re-suspended with 5 mL of Epstein-Barr virus (EBV) media from the EBV producer cell line B95.8 and 2 mL Cyclosporine-A solution (at 4 μg/mL; Sigma), transferred to a T25 flask, and placed into a CO2 incubator (5% CO2, 37°C, ∼85% humidity). After 7 days, growing cells were split into additional flasks once they reached 75% confluence. The lymphoblastoid cell lines were cultured and maintained at the OSUCCC Biospecimen Services Shared Resource.
Gene expression analysis
Total RNAs from lymphoblastoid cell lines obtained from family members and controls (7 with the TINF2 mutation, and 20 controls with wild-type TINF2) were extracted by using Trizol solution according to the manufacturer's protocol (Invitrogen). The reverse transcription-polymerase chain reactions (RT-PCRs) were performed by using SYBR Green assays. Glyceraldeyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous control. Quantitative real-time PCR was performed by using a 7900HT Fast Real-Time PCR System (Applied Biosystems). The formula 2−ΔCt, where ΔCt = Ct(GENE) − Ct(GAPDH) was employed to calculate the relative transcript levels. Primers for amplification of TINF2 and GAPDH are listed in Supplementary Table S1.
Making expression constructs, cell culture, and transfection
Expression plasmid DNA constructs of TINF2 and TERF1 were purchased from Origen (Cat No.: RC214419 and RG217957, respectively; OriGene Technologies, Inc.). Mutant TINF2 (
Co-IP and Western blot
co-IP was performed by using Pierce c-Myc-Tag Magnetic IP/Co-IP Kit (Cat No.: 88844; Thermo Scientific) according to the manufacturer's protocol. Briefly, cells were washed twice with PBS and they were lysed by using Mag c-Myc IP/Co-IP lysis buffer (buffer-1). Protein concentration was measured by using the Pierce BCA Protein Assay Kit (Cat No.: 23227; Thermo FisherScientific). Equal protein amounts of each sample were added to prewashed c-Myc magnetic beads and incubated for 30 minutes at room temperature with rotation. The beads were collected with a magnetic stand and washed three times with buffer-2 from the kit. Proteins were eluted from the beads with nonreducing samples buffer (provided by the kit).
Western blot was performed by using the Bio-Rad mini PROTEAN TGX Gel (Cat No.: 4568093; BioRad) and the Transfer Pack (Cat No.: 1704158; BioRad). Primary antibodies of anti-Flag (Cat No.: F7425, 1:1000; Sigma) and anti-TERF1 (Cat No.: HPA048379, 1:1000; Sigma) and the secondary antibody of anti-rabbit IgG (Cat No.:7074S, 1:5000; Cell Signaling) were used. Input loading control was done by using anti-GAPDH antibody (sc-20357, 1:1000; Santa Cruz). The LI-COR Odyssey Fc Imagine System was used to detect the chemiluminescence.
Measurement of relative telomere length
Genomic DNAs extracted from lymphocytes (whole blood samples) were used. The quantitative polymerase chain reaction (qPCR) assay was performed as previously described (20). Briefly, the sequences of PCR primers for the telomeres were: Forward, 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT; Reverse, 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT. Primers for the 36B4 gene: Forward 5′-CAGCAAGTGGGAAGGTGTAATCC; Reserve, 5′-CCCATTCTATCATCAACGGGTACAA. All PCRs were performed by using the PowerUp SYBR Green master mix (Cat No.:A25742; Applied Biosystem). The thermal cycling profile for the telomere amplification consisted of 95°C 5 min, followed by 40 cycles of 95°C for 15 s and 56°C for 30 s. After amplification, a melting curve was created to confirm the specificity of the reaction.
Relative telomere length (RTL) was quantified in terms of the ratio of telomere repeat copy number “T” to single copy gene copy number “S” (T/S-ratio). We used the 36B4 (encoding acidic ribosomal phosphoprotein P0) as the control single copy gene. All samples, including the endogenous control, were run on the same 384-well plate to eliminate the effect of inter-assay variability.
WES and data analysis
WES of 14 NMTC patient DNA samples from 7 families (2 individuals per family) was performed by Novogene Co. Ltd., CA using the HiSeq X (Illumina) platform. After cleaning read pairs by removing adapter contamination and reads with uncertain and low-quality bases, Burrows–Wheeler Aligner was used to map the amplified sequences to the human reference genome. Picard (
Variant filtering and annotation
Variants in the coding regions of the 6 shelterin genes (TINF2, TERF1, TERF2, ACD, TERF2IP, and POT1) in 24 families were annotated by ANNOVAR (23). Only the variants shared by two or more affected individuals in at least one family were selected. A cutoff value of 0.05 was used for the maximum allele frequency in the gnomAD, Exome Sequencing Project, Exome Aggregation Consortium, 1000 Genomes, TCGA, and COSMIC databases. Predicted functional consequences of the missense variants were annotated by using SIFT, PolyPhen-2, and Variant Effect Predictor (VEP).
Databases and web resources
gnomAD:
NHLBI GO Exome Sequencing Project (ESP):
The Exome Aggregation Consortium (ExAC):
The 1000 Genomes Project:
The Cancer Genome Atlas (TCGA):
Oncomine database:
The Catalogue of Somatic Mutations in Cancer (COSMIC):
SIFT:
PolyPhen-2:
VEP:
Statistical analysis
We used nonparametric tests to compare RTLs in family members with the TINF2 mutation and those without the mutation and other controls. The Kruskal–Wallis test was used for comparing three groups, and the Tukey–Kramer tests were used for pairwise comparison. Gene expression analysis was performed by applying linear mixed models. All the analyses were two sided, and a p-value of <0.05 was considered statistically significant.
Data availability statement
The WGS data were previously reported (11). The WES sequence data are not publicly available, because the data contain information that could compromise research privacy/consent.
Results
A rare germline TINF2 frameshift mutation in a large PTC and melanoma family
The key family in this study is a large three-generation family with individuals affected with PTC and melanoma and other diseases (Fig. 1 and Table 1). In our recent WGS study of 17 NMTC families, we identified an ultra-rare frameshift variant caused by a single nucleotide deletion in this family [C/−, chr14:24,710,239 (GRCh37.p13), NM_012461.3:c.591delG, p.Gly197fs, in Family I] (11). According to the recommended mutation nomenclature by the Human Genome Variation Society, we renamed this frameshift mutation as
This germline mutation is novel and rare. It has not been reported in the gnomAD datasets spanning 125,748 exome sequences and 15,708 whole genome sequences from unrelated individuals and to the best of our knowledge not in any other populations or somatic mutation databases. This family is the same one reported in an earlier study (19). In our first genome-wide linkage analysis, at least four prominent linkage peaks (8q24, 12q21–24, 14q11, and 6q27) were identified (19). We re-performed genome-wide linkage analysis with additional samples from this family (11). The four prominent linkage peaks described earlier remained unchanged. The 14q11 locus spans about 4.2 Mb, covering the genomic region of the TINF2 gene. In this study, we further examined the TINF2 mutation in all samples available from this family, including eight individuals affected with PTC and/or melanoma, four individuals with benign thyroid diseases, one individual with CLL, and one unaffected individual who is the spouse of the affected individual No. 1 (Fig. 1). The clinical characteristics of these individuals are shown in Table 1. We confirmed that the TINF2 mutation is only present in the individuals affected with PTC and/or melanoma (designated as affected), and absent in all the other six individuals (designated as unaffected) (Fig. 1 and Table 1). These data indicate that the TINF2 mutation completely co-segregates with the PTC and melanoma cancer types, but not with benign thyroid diseases or CLL in this family.
Effects of the TINF2 mutation on gene expression and interaction with TERF1
The most abundantly expressed TINF2 transcript in thyroid and skin tissues and lymphoblastoid cells is the TINF2 isoform ENST00000399423.4 (RefSeq NM_012461.3) according to the GTEx database (
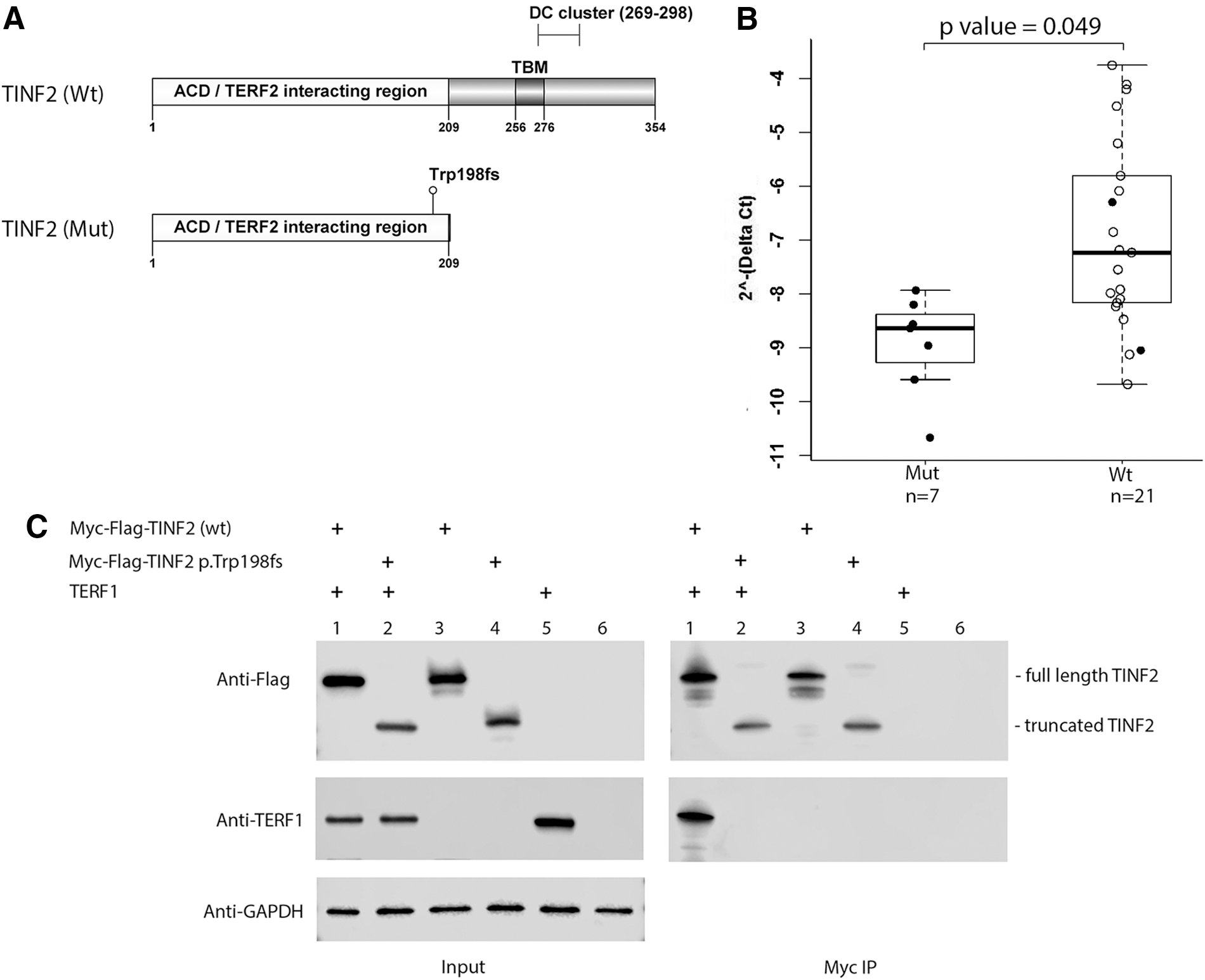
Analyses of the mutant TINF2. (
Since the TBM motif is critical for TINF2 binding to TERF1 (12,23), we reasoned that the truncated TINF2 could be lost or have damaged binding ability to TERF1. To address this question, we made expression constructs of mutant and wild-type TINF2. Western blot of cell lysates from transfected cells revealed stable expression of truncated and wild-type TINF2 in the HEK293T cell line (Fig. 2C). To assess the interaction between TINF2 and TERF1, we co-transfected expression constructs of TERF1 with either wild-type or mutant Myc-FLAG-TINF2, followed by co-IP assay and Western blot. The data shown in Figure 2C indicate that no detectable TERF1 binding to mutant TINF2 was observed, suggesting that TINF2
Longer telomere length in the mutant patient DNA from lymphocytes
Due to the role of TINF2 in telomere maintenance, we hypothesized that the mutant TINF2 could affect telomere length. We assessed RTL by using a qPCR assay with DNA samples from whole blood from eight affected individuals who carry the TINF2 mutation and six unaffected individuals who carry the wild-type TINF2 (Fig. 1). All subjects except one (individual No. 18, melanoma, age 26 at diagnosis) are within the age range of 50 to 84 years old (Table 1). We also assessed RTLs in age-matched Ohio healthy controls (n = 62) (median age of 61, age range of 50 to 84, 25 male and 37 female). The comparison of the RTLs is shown in Figure 3. For the purpose of matched age range, we excluded individual No. 18 from this analysis. There is a significant difference in RTLs among the three groups with a p-value of 0.005. The RTLs are generally longer in the affected individuals with the mutant TINF2 as compared with individuals with the wild-type TINF2 in the family. The median RTL of the mutant TINF2 group is significantly longer than that of the wild-type group and the control group with p-values of 0.031 and 0.004, respectively. There is no difference between RTLs in family members with wild-type TINF2 and the controls (p = 0.958) (Fig. 3).
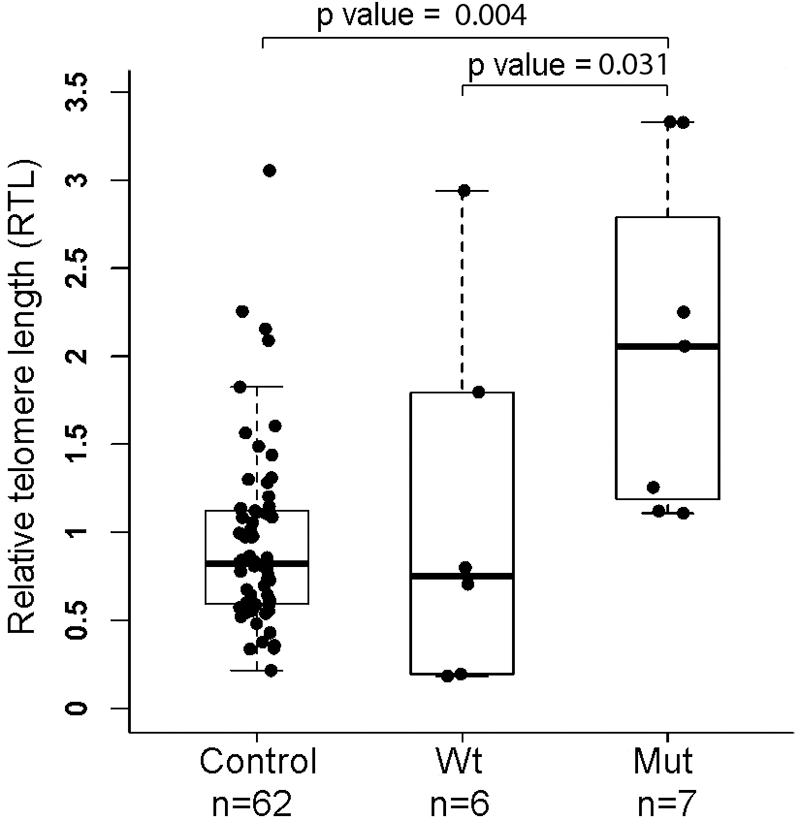
Measurement of RTL. RTLs were measured by using a quantitative PCR assay with blood DNAs from family members with or without the TINF2 mutation and healthy controls. N, sample size. Nonparametric tests (the Kruskal–Wallis test for comparing three groups and the Tukey–Kramer test for pairwise comparison) were performed. The RTL experiments were repeated for three times. RTL, relative telomere length.
The RTL of individual No. 18 was 1.50, which is longer than the median RTL (0.82) of the control group. The measurement of RTLs was further validated by using a different primer set in the qPCRs; we obtained completely consistent results (Supplementary Fig. S2).
Germline coding variants in six shelterin genes in NMTC families
We screened for germline coding variants in the 6 shelterin genes (TINF2, TERF1, TERF2, ACD, TERF2IP, and POT1) in a total of 24 families by using the WGS data from 17 families as described by Wang et al. (11) and the WES in 7 new families (Supplementary Fig. S1). In these seven families, there are at least three individuals affected with PTC and some individuals affected with melanoma and other types of cancers. Here, we considered only rare coding variants with minor allele frequency of <0.05 and the variants shared by the two or more affected individuals in each family. Three missense variants (two in the TINF2 gene and one in the ACD gene) and one synonymous variant in the TERF2IP gene were identified. Functional annotation of the three missense variants was performed by using software tools or web servers of SIFT, PolyPhen-2, and VEP (Table 2 and Supplementary Table S2). The variants in the TINF2 gene (SNPs rs1464083474 and rs202093758) were predicted as “tolerated” or “benign.” The variant in the ACD gene (SNP rs142662151) was predicted as “deleterious” or “possibly damaging.”
Three Missense Variants in Shelterin Genes in 24 Nonmedullary Thyroid Cancer Families
Genomic position: GRCh37.p13.
The families B and H and the WGS data have been reported (11).
ALT, altered allele; REF, reference allele; WGS, whole genome sequencing.
Discussion
The TINF2 mutation is a novel germline DNA variant detected in individuals with PTC and/or melanoma in a large family. It is not reported in any population databases we searched, and not found in any other type of cancer, including sporadic thyroid cancer or melanoma. TINF2, the central component of shelterin, is a hub that interacts with TERF1, TERF2, and ACD by mediating the assembly of the entire complex (26,27). The truncating TINF2
Here, we found that the truncated TINF2
Telomeres play important roles in cell division, genomic instability, and the initiation of tumorigenesis. Both the telomerase and shelterin complexes are involved in telomere maintenance. In human somatic cells, telomeres shorten gradually with each cell division, and cells with extremely shortened telomeres reach senescence or apoptosis. However, cancer cells have aberrant mechanisms of telomere maintenance: Their telomeres are shortened, but frequently not short enough to lead to cell senescence and therefore acquire immortality (18,32,33). Interestingly, investigations into the relationship between RTL and risk of cancer lead to paradoxical observations. Multiple studies have observed that shorter telomeres are associated with increased risk for most cancers; however, in the general population, individuals with constitutively long telomeres are at a higher risk for major cancer (18,34
–36). We observed that the TINF2
TINF2 is a subunit of the shelterin complex; this complex protects telomere ends and cooperates with telomerase to maintain telomeres. TINF2 is involved in the regulation of telomere length by connecting the double-stranded DNA-binding proteins TERF1 and TERF2 to the single-stranded DNA-binding protein ACD (14,27). Besides its role in stabilizing shelterin and in regulating tankyrase action, TINF2 might regulate telomerase by affecting the higher-order structure of mammalian telomeres (29,37,38). TINF2 was previously shown to be a negative regulator of telomere length (12,38). When TINF2 is inhibited with mutant alleles or RNAi, telomeres become overelongated by telomerase, whereas overexpression of TINF2 inhibits telomere elongation in human cell lines (12,38). In line with our data, genome-wide association in thyroid cancer identified DNA variants associated with telomere length. The alleles of SNPs rs6793295 on 3q26 and rs7902587 on 10q24 are associated with longer telomeres in NMTC patients (10). These data suggest that longer telomeres could be a predisposing factor for thyroid cancer as well as for melanoma. We hypothesize that the TINF2
Germline mutations have been identified in the TINF2 gene in autosomal dominant inheritance of short telomere syndromes, such as dyskeratosis congenita (DC), Revesz syndrome, Hoyeraal-Hreidarsson syndrome, and pulmonary fibrosis (39). These diseases are characterized by presenting with critically short telomeres (39
–41). TINF2 mutations in these patients map to a small region (amino acids 269–298) labeled DC-cluster in Figure 2A. The most common mutations are missense mutations found at amino acids 280–282, but nonsense mutations have also been reported (39,42). While the most common mutation TINF2
We have previously reported a long noncoding RNA gene (PTCSC1) in 8q24 as a candidate predisposing gene in the same thyroid cancer family described in this article (19). The risk haplotype of the PTCSC1 gene is a common haplotype among the PTC families we examined; however, the function of the PTCSC1 gene remains unknown (19). The presence of at least two risk alleles of PTCSC1 (in 8q24), and TINF2 (in 14q11) suggests a polygenic pattern of inheritance in thyroid cancer. It is unclear whether the two genes can act in cooperation or through shared common pathways. Further research is needed to elucidate the exact pathophysiology and genetic pathways in thyroid cancer predisposition.
Germline mutations/variants in the shelterin genes POT1 and ACD have been implicated in familial melanoma, familial glioma, Li-Fraumeni-like syndrome, mantle cell lymphoma, and CLL (44 –50). To explore what other DNA variants in the shelterin genes might confer thyroid cancer risk, we screened for coding variants in the six shelterin genes (TINF2, TERF1, TERF2, ACD, TERF2IP, and POT1) by using WGS data from 17 families and WES data from 7 families. Overall, coding variants in the shelterin genes turned out to be rare in these families. The functional significance of these variants will be further explored in our future work.
In summary, a rare frameshift mutation in the TINF2 gene co-segregates fully with PTC and melanoma, but not with benign thyroid diseases or CLL in a large family. The truncated TINF2
Footnotes
Acknowledgments
The authors thank Jan Lockman and Barbara Fersch for administrative help, and the OSUCCC Genomics Shared Resource and Biospecimen Services Shared Resource.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by National Cancer Institute Grants P30CA16058 and P01CA124570.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2