
Abstract
Background:
Pediatric thyroid cancer has characteristics that are distinct from adulthood thyroid cancer. Due to its very low prevalence, little is known about the genetic characteristics of pediatric follicular thyroid cancer (FTC).
Methods:
We investigated genetic alterations in tumor tissues from 15 patients aged <20 years (median: 14.3 years; range: 2.4 − 19.0 years) using multifaceted approaches. Whole-exome sequencing, targeted next-generation sequencing using a cancer gene panel, and Sanger sequencing of the major exons of the H/K/N-RAS and DICER1 genes and the promoter region of the TERT gene were performed. Normal tissues or blood of patients with DICER1- or PTEN-positive tumors was also evaluated to determine whether the variant is germ line.
Results:
The median tumor size was 3.1 cm (range: 0.6 − 6.4 cm). Four patients exhibited angioinvasion and one extensive capsular invasion; none showed evidence of disease over a median of 8.1 years. Eight patients (53.3%) had DICER1 variants, including four with DICER1 syndrome (three patients were <10 years of age). One patient had a germ line PTEN frameshift variant with the diagnosis of PTEN hamartoma tumor syndrome. One patient had a PAX8/PPARγ rearrangement, and two patients had no genetic driver alteration other than multiple loss of heterozygosity with or without copy number alterations in their tumors. No RAS or TERT variants were found. Nodular hyperplasia and follicular adenoma (FA) coexisted in DICER1 variant-positive FTCs more frequently than variant-negative FTCs (p = 0.026). All DICER1 variant-positive FTCs had a somatic missense variant at metal binding sites (six at codon p.E1813 and two at codon p.D1709) within the RNase IIIb domain; seven had other missense, nonsense, or frameshift variants in the DICER1 gene. Six coexisting FAs of two patients with DICER1 syndrome (three of each) had additional somatic variants at metal binding sites within the RNase IIIb domain (codon p.E1705, p.D1709, p.D1810, or p.E1813), different from each other and from the indexed FTC tumor.
Conclusions:
Pediatric FTCs have distinct genomic alterations and pathogenesis compared with adults, particularly those characterized by DICER1 variants. The DICER1 variant should be considered in pediatric FTCs, especially in cases <10 years of age. In all DICER1 variant-positive FTCs and FAs, recurrent hotspot variants were found at metal binding sites within the RNase IIIb domain, suggesting they impact tumorigenesis.
Introduction
Thyroid cancer is uncommon in pediatric populations. Pediatric thyroid cancer differs from adulthood thyroid cancer in clinicopathological characteristics and prognosis (1). The link between childhood exposure to radiation fallout and the risk of thyroid cancer in adolescents in Chernobyl has been well documented (2), and the genomic landscape of post-Chernobyl radiation-induced thyroid cancers in children has been reported (3). Most genetic studies have been conducted in papillary thyroid cancer (PTC) cases (4 –10), with gene fusions found in a higher percentage of pediatric patients compared with the single-nucleotide variants (SNVs) frequently reported in adult patients (11).
This provides insight into the difference in the spectrum of genomic alterations and pathogenesis between pediatric and adult patients with histologically similar thyroid cancers (12).
Follicular thyroid cancer (FTC) is relatively uncommon during childhood, accounting for 5–10% of pediatric thyroid cancers (1,13). Pediatric FTC cases in turn account for 1.9% of all FTC cases, according to data from the National Cancer Institute Surveillance, Epidemiology and End Results (14) and a Japanese database (15). Although some studies reported that pediatric FTC patients exhibited poorer prognoses than those diagnosed between 20 and 45 years of age (16), prognoses similar to those of adult FTC patients aged ≥21 years have also been reported (15). The most frequently detected genetic alterations in adult Korean FTC cases are H/K/NRAS variants (30–50% of all such alterations), and PAX8-PPARγ rearrangements (∼2%) (17,18). However, little is known about the genetic characteristics of pediatric FTC patients. A previous study reported an NRAS variant and a PAX8-PPARγ fusion in five pediatric FTC cases (19). Another recent study, of 41 patients aged <21 years, reported a low prevalence of N/H/KRAS variants (12.2%), and the absence of PAX8-PPARγ fusions (20). Both studies evaluated a small number of candidate genes using direct (or pyro-) sequencing or a reverse transcription-polymerase chain reaction (RT-PCR) method. More comprehensive genetic assessments are therefore needed to enhance our understanding of the molecular characteristics of pediatric FTC patients.
In this study, we aimed to identify genetic alterations of pediatric FTCs using next-generation sequencing (NGS) methods. We also evaluated associations with the prognoses and clinicopathological characteristics of pediatric FTC patients according to genetic alterations.
Materials and Methods
Patients
In total, 17 (9.3%) of 182 thyroid cancer patients younger than 20 years, who underwent surgery at Seoul National University Hospital (SNUH, Seoul, Republic of Korea) between 1983 and 2018, were diagnosed with FTC. After excluding two patients for whom tumor tissue samples were not available, 15 patients (3 males and 12 females; median age: 14.3 years; range: 2.4–19.0 years) were finally included in this study. All patients were retrospectively reviewed for clinicopathological characteristics and prognoses, and assessed for personal or family history of syndromic cancers. This study was approved by the Institutional Review Board (IRB) of SNUH, in accordance with the Declaration of Helsinki (approved ID: H-1707-034-501 and 1505-023-670). Written informed consent was obtained in line with our IRB protocol.
Treatment and follow-up strategies
The treatment and follow-up strategies used in this study were reported previously (1). In brief, all patients underwent total thyroidectomy (TT), subtotal thyroidectomy, or lobectomy. Prophylactic central node dissection was introduced in 2003 and has been performed in most patients with PTC ≥1 cm since 2007 in our hospital. The presence of lung metastasis was detected by radiological examination (i.e., 131I whole-body scan [WBS] and/or chest computed tomography [CT]). Postoperative 131I therapy was recommended for patients with a large tumor (>1 cm), locoregional and/or lung metastasis, and the timing and number of 131I doses were individualized. After the initial treatment, patients were monitored by clinical examination, serum thyroglobulin (Tg) and anti-Tg antibody (TgAb) levels, as well as neck ultrasonography. CT, 131I WBS, and positron-emission tomography were performed if indicated.
Definition of disease outcome
In the initial dynamic risk stratification, the patients' response to treatment was classified as excellent, indeterminate, biochemical incomplete, or structural incomplete based on their serum Tg and TgAb levels and imaging study findings during the first two years of follow-up (21). The final disease outcomes were categorized as no evidence of disease (NED), persistent disease (biochemical or structural), and recurrent disease based on findings at the last follow-up visit (22).
Blood and tissue samples
To exclude familial-syndrome-associated nonmedullary thyroid cancer (23), blood samples were drawn from patients (n = 9) harboring a DICER1 or PTEN variant in tumor tissue, and from the parents of those harboring germ line DICER1 variants (n = 4); leukocyte DNA was isolated using the RecoverAll™ Total Nucleic Acid Isolation Kit (Life Technologies, Carlsbad, CA). Formalin-fixed, paraffin-embedded (FFPE) tissue samples were collected and categorized according to the histology report as normal thyroid, nodular hyperplasia (NH), follicular adenoma (FA), or FTC. Pathological diagnoses were made according to the latest World Health Organization classifications for thyroid cancer (24). All FFPE samples were reviewed by one expert pathologist (K.C.J.) (Supplementary Fig. S1), and lesions were manually dissected under a microscope to a thickness of 4 μm. The American College of Medical Genetics and Genomics (ACMG) guidelines were used to classify germ line variants into a five-tier pathogenicity status (25,26).
Fluorescence in situ hybridization
Analyses of PAX8/PPARγ rearrangements in FFPE tumor tissues were performed by fluorescence in situ hybridization using the PPARγ (3p25) dual-color break probe (Kreatech Diagnostics, Amsterdam, The Netherlands), as described previously (18).
Targeted or whole-exome sequencing
DNA samples from FFPE tissues of 14 of 15 FTC tumors (except one with a previously confirmed PAX8/PPARγ rearrangement) were submitted to targeted or whole-exome sequencing (WES) at Macrogen (Seoul, Republic of Korea) or at SNUH (Supplementary Table S1).
Targeted sequencing was performed using either the SNUH FIRST cancer panel, version 3 (SNUH) or a customized thyroid cancer panel (Macrogen) (18). Both panels used a hybridization capture probe based on the SureSelect Target Enrichment method (Agilent Technologies, Santa Clara, CA). The target genes of both panels are listed in Supplementary Table S2. The targeting (sequencing) probe covered the intronic region, in which structural variations (SVs) frequently occur, and the TERT gene promoter region in addition to the exonic regions of cancer-related genes. WES libraries were prepared using the SureSelectXT Library Preparation Kit (SureSelect Human All Exon V6; Agilent). All libraries were sequenced by the HiSeq platform (Illumina, San Diego, CA) as paired-end reads. Sequencing reads were aligned to the reference human genome (hg19) using the Burrows/Wheeler aligner (27). Processing continued until the “BAM” file was created following the Genome Analysis Toolkit (GATK) best practice recommendations using SAMtools (28), Picard and GATK (29). Analysis to detect SNVs, insertions/deletions (indels), copy number alterations (CNAs), and SVs followed the method of the FIRST cancer panel. CNA analysis was restricted to data derived from the WES and FIRST cancer panel, because the thyroid cancer panel had an insufficient bed size (786 Kb) to detect CNA. Considering the probe density, CNA detection was limited to more than 10 Mb and arm level in the WES and FIRST cancer panel, respectively.
RAS, DICER1, and TERT sequencing
Samples of the FFPE tissue block were digested with proteinase K (Sigma-Aldrich, St. Louis, MO) for more than 24 hours at 56°C, and DNA was isolated from the digested tissue using a Tissue SV Mini kit (General Biosystem, Seoul, Republic of Korea). PCR was performed using a BioMix kit (Bioline, Taunton, MA). The TERT promoter (C228T and C250T) region was amplified in all 15 patients using PCR with appropriate primers (18) (Supplementary Tables S3, S4, S5). For three FTC samples that failed to yield NGS libraries and one sample with a PAX8/PPARγ rearrangement, frequently mutated regions of H/K/NRAS (codon 12/13 and codon 61) and DICER1 were amplified using PCR with appropriate primers (Supplementary Tables S3, S4, S5). Purification and Sanger sequencing of the PCR products were conducted by Macrogen. In all cases with DICER1 or PTEN variants in the FTC tumor sample, the presence of germ line DICER1 or PTEN variants was evaluated in leukocyte DNA. In addition, if tumors other than the primary FTC existed, all possible tissue samples, including normal thyroid, NH, and FA, were evaluated for somatic DICER1 variants. Each DNA sample was assayed at least twice to confirm the variant status.
Statistical analysis
The Mann–Whitney U-test was used to compare non-normally distributed continuous variables between two groups. Categorical variables were compared using Fisher's exact test. A value of p < 0.05 was considered statistically significant in the multivariate models. All analyses were performed using SPSS statistical software for Windows (version 25.0; SPSS, Inc., Chicago, IL).
Results
Clinicopathological characteristics of 15 pediatric FTC patients
Genetic alterations and clinicopathological characteristics of 15 FTC patients are shown in Table 1. Of the 15 patients, 13 (86.7%; all except P11 and P13) underwent TT, and four (26.7%) underwent prophylactic central lymph node dissection. The tumor subtypes were minimally invasive FTC (miFTC) (n = 10), encapsulated angioinvasive FTC (eaFTC) (n = 3), and widely invasive FTC (wiFTC) (n = 2). The overall median tumor diameter was 3.1 cm (range: 0.6–6.5 cm). Angioinvasion was detected in four patients, one of whom showed extensive capsular invasion. In total, seven (46.7%) received 131I therapy in the postoperative period. No patient had tumors with extrathyroidal extension, or lymph node or lung metastasis. Patients were followed up for a median duration of 8.1 years (range: 0.5–13.4 years). The nine patients who were followed for more than two years had an excellent response to the initial treatment during the first two years of follow-up, and NED based on serial Tg and radiological tests at the last follow-up visit.
Clinicopathologic Characteristics at Presentation and Prognosis in Follicular Thyroid Cancer Patients Diagnosed <20 Years of Age
Pathological N (pNx) staging was determined by lymph node findings, and clinical N staging (cNx) was assessed using biochemical and imaging findings.
M staging was based on cross-sectional imaging such as whole-body scan, chest computed tomography, or positron-emission tomography.
In the initial DRS, the patients' response to treatment was classified as excellent, indeterminate, biochemical incomplete, or structural incomplete based on their serum Tg and anti-Tg antibody levels and imaging study findings during the first two years of follow-up.
Loss of heterozygosity with or without copy number gain/loss.
All negative for RAS, TERT, and DICER1 variants at Sanger sequencing in three patients who failed to construct next-generation sequencing library.
Bilateral lesion.
DRS, dynamic risk stratification; eaFTC, encapsulated angioinvasive FTC; FA, follicular adenoma; FTC, follicular thyroid cancer; F/U, follow-up; LOH, loss of heterozygosity; miFTC, minimally invasive FTC; NED, no evidence of disease; NH, nodular hyperplasia; N/I, not identified; ; Tg, thyroglobulin; wiFTC, widely invasive FTC.
Genomic alterations and clinicopathological characteristics of pediatric FTCs
Eight (53.3%) of 15 patients had tumors with DICER1 variants (cases P1 to P8; Table 1; Supplementary Table S6); four (P1–3 and P5) had a germ line variant, and two showed loss of heterozygosity (LOH) with or without copy number gain or loss at multiple chromosomes (P5 and P8; Supplementary Fig. S2A). One patient had a pathogenic germ line PTEN frameshift variant (P9). One patient had a tumor with a PAX8/PPARγ rearrangement (P10). Multiple LOH with or without CNAs was found in five FTCs; two patients showed no other coexisting variant (P11 and P12; Supplementary Fig. S2B), while three had other variants (P5 with germ line and somatic DICER1 variants and P8 with somatic DICER1 variants, as described above, and P9 with a germ line PTEN variant; Supplementary Fig. S2B). The remaining three patients (P13–15), for whom we failed to construct an NGS library, were all negative for RAS and DICER1 variants based on Sanger sequencing (Table 1). TERT promoter or RAS variants were not present in any of the 15 tumors investigated.
All eight DICER1 variant-positive FTCs had a somatic missense variant involving metal binding glutamate or aspartate residues in the RNase IIIb domain (six at codon p.E1813 and two at codon p.D1709). Additional missense, nonsense, or frameshift variants in DICER1 were found in seven of the eight FTCs; three were somatic variants and four were pathogenic germ line variants (Fig. 1 and Table 2; Supplementary Table S6). Eventually, four patients (P1–3 and P5) were diagnosed with DICER1 syndrome. One patient with a pathogenic germ line PTEN frameshift variant (P9) was diagnosed with PTEN hamartoma tumor syndrome (Cowden syndrome). LOH was identified in chromosomes 7 and 16 without additional variants in her tumor. She had coexisting manifestations, including recurrent breast fibroadenomas, arteriovenous malformations, and hemangiomas in the brain.
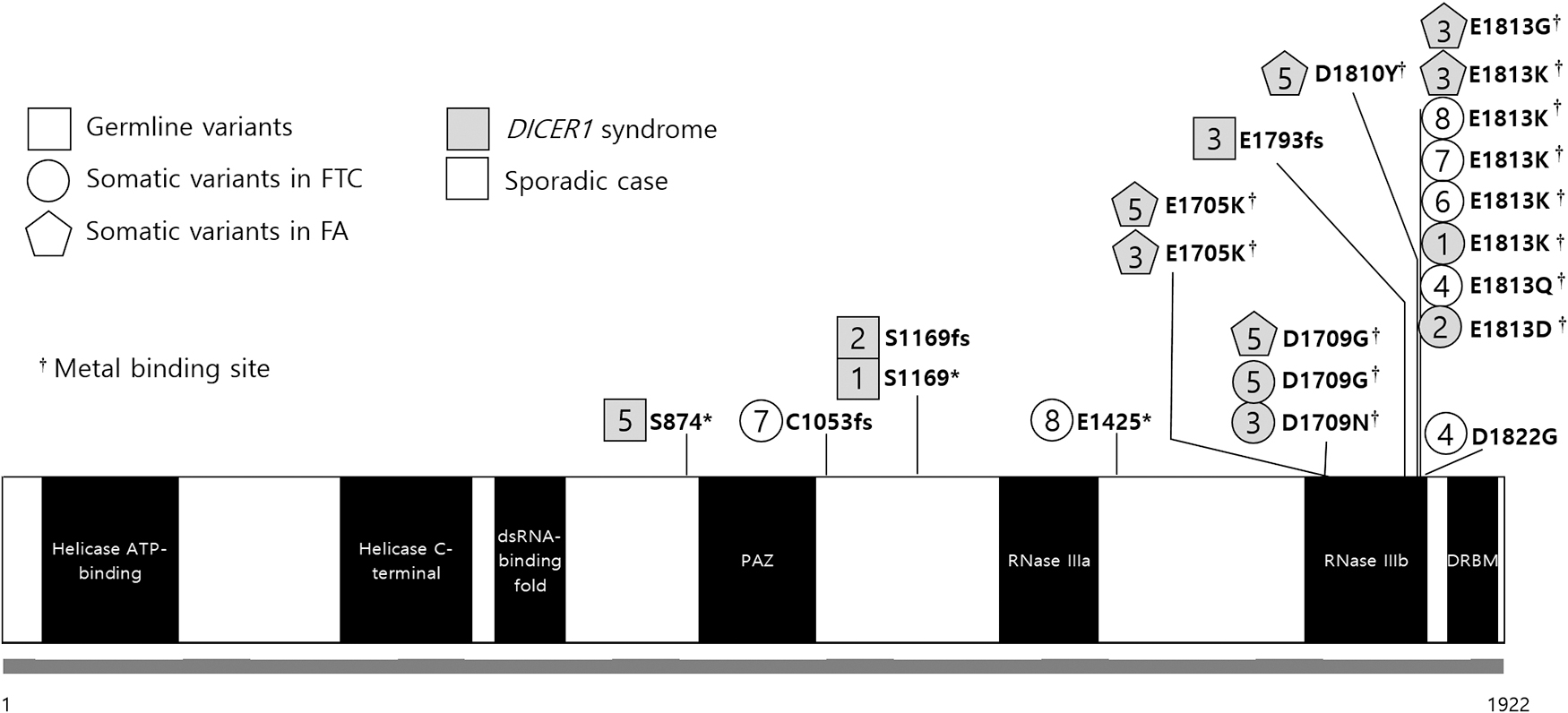
Diagram of the DICER1 variants and their positional relationship with the RNase IIIb domain. Case numbers are indicated by squares (germ line variants), circles, or pentagons (somatic variants). Gray-filled areas represent the DICER1 syndrome patients (P1–3 and P5). The germ line truncating variants are widely distributed in the DICER1 gene, and the somatic missense variants of the DICER1 syndrome cases are located at codon p.D1709 or p.E1813. FA, follicular adenoma; FTC, follicular thyroid cancer.
Follicular Thyroid Cancer Patients with DICER1 Syndrome
ACMG, The American College of Medical Genetics and Genomics; MNG, multinodular goiter; N/A, not available; PNET, primitive neuroectodermal tumor; PPB, pleuropulmonary blastoma.
When clinicopathological characteristics were compared between DICER1-positive (n = 8) and DICER1-negative (n = 7) patients, NH and/or FA coexisted more frequently in DICER1 variant-positive FTCs (p = 0.026 using the Fisher exact test). All FTC patients aged <10 years had DICER1 variant-positive tumors. Otherwise, no differences were found in sex, age, tumor size, pathological findings, or prognoses between the two groups (Table 1).
Molecular and histological characteristics of eight DICER1-positive tumors
The four patients with DICER1 syndrome all presented with a combination of a truncating germ line DICER1 variant and a tumor-specific hotspot missense variant. While the germ line truncating variants were widely distributed in the DICER1 gene (p.S874*, p.S1169*, p.S1169fs, and p.E1793fs), the somatic missense variants were located at codon p.D1709 or p.E1813. The other four patients lacking germ line DICER1 variants had two (P4, P7, and P8) or one (P6) somatic DICER1 variant(s) in tumor tissue; all four patients had a variant in the same codon (p.E1813), and three had additional variants (p.D1822G, p.C1753fs, and p.E1425*) at different positions (Fig. 1 and Supplementary Table S6).
For two patients (P3 and P5) with DICER1 syndrome and multinodular goiters, FFPE tumor samples at different stages (normal, NH, FA, and FTC) were available. In patient 3 (P3), five well-demarcated nodules (two in the right lobe and three in the left lobe) were found in the resected thyroid specimen (Fig. 2A). The FTC in the right lobe, which was surrounded by a thick and irregular fibrous capsule, showed microfollicular proliferation and capsular invasion of tumor cells. Three FAs (one in the right lobe and two in the left lobe) showed a mixed microfollicular and normofollicular pattern, and were encapsulated by a thin fibrous band. The tumor cells of these four lesions (one FTC and three FAs) had round and hyperchromatic nuclei. However, nuclei of the follicular cells in the other hyperplastic nodule in the left lobe were less hyperchromatic compared with the FTC and three FAs, and a fibrous capsule was not evident. In patient 5 (P5), four nodular lesions (three in the left lobe and one in the right lobe) were seen (Fig. 2B). The FTC and FA in the left lobe showed microfollicular proliferation with a focal papillary pattern, although nuclei were round and papillary carcinoma-like nuclear features were not seen. The tumor cells of the FTC invaded the adjacent normal parenchyma. The other FAs (one in the left lobe and one in the right lobe) showed mixed microfollicular and normofollicular patterns.
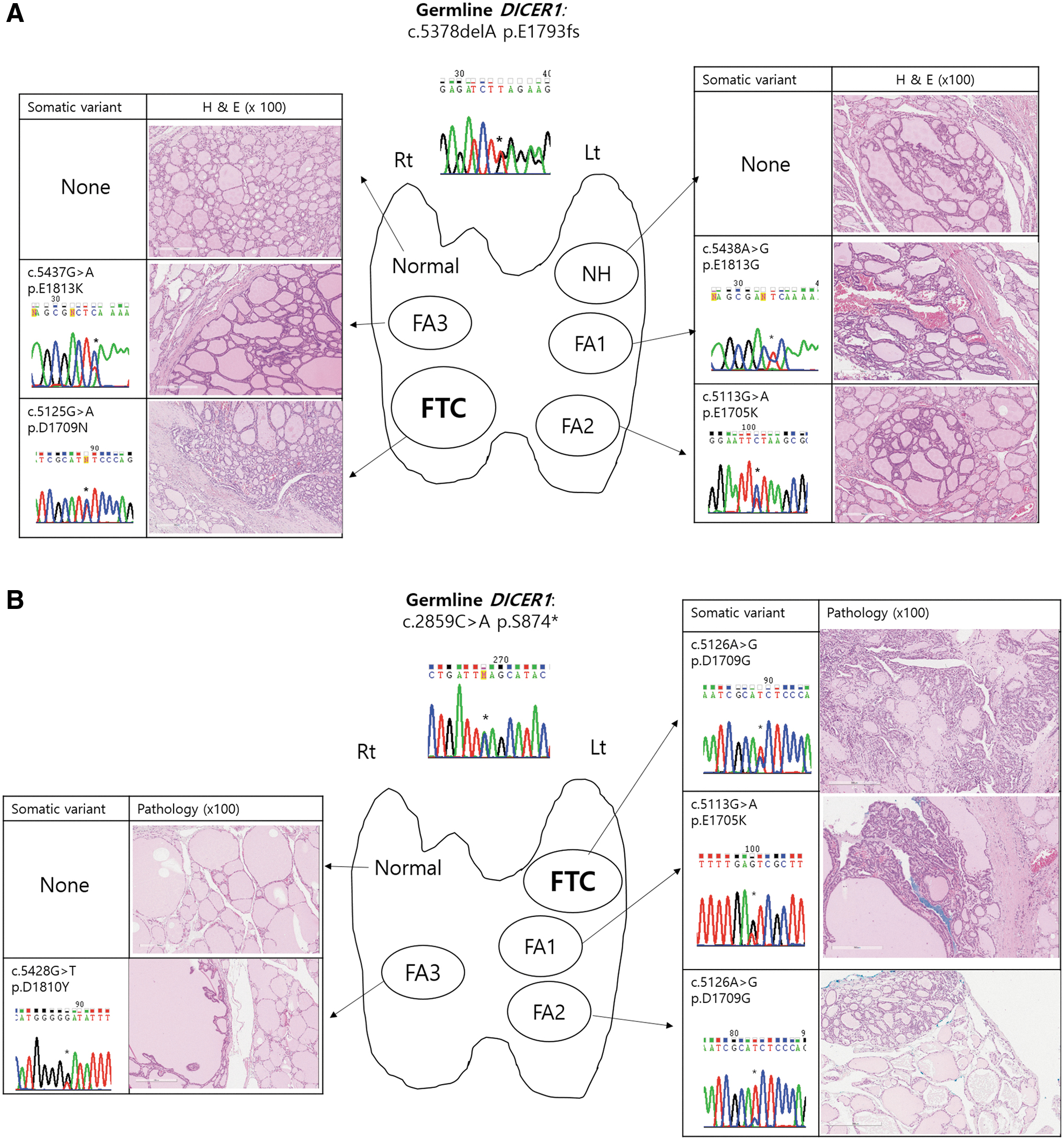
Genetic alteration and histology of normal thyroid, nodular hyperplasia, follicular adenoma, and follicular thyroid carcinoma in two patients with DICER1 syndrome presenting with multinodular goiters (P3 in
We evaluated the genetic alterations of the DICER1 gene in normal, NH, and/or FA tissues and compared the results with the index FTC data. Notably, even within the same patient, tumors differing in histology or site had different DICER1 variants; all six coexisting FAs of two patients had additional somatic variants in codon p.E1705, p.D1709, p.D1810, or p.E1813, in addition to the germ line truncating variants, which were different from each other and from the indexed FTC tumor (Figs. 1 and 2). However, no additional variant was found in the tissues of the normal thyroid and NH (Fig. 2). These results suggested that variants at metal-binding sites in the RNase IIIb domain played important roles during the development of FAs or FTCs.
Clinical characteristics of four patients with DICER1 syndrome
All four patients with DICER1 syndrome (P1–3 and P5; Table 2) had a detectable FTC because of a palpable neck mass. A family history of multinodular goiters (n = 2) and/or a history of an embryonal tumor (n = 2) were associated with DICER1 syndrome. Two patients (P3 and P5) had one or more family member(s) with multinodular goiters and other coexisting FAs and/or NH, compatible with familial multinodular goiter. Three patients (P2, P3, and P5) had first- or second-degree relatives who had undergone thyroidectomy due to a benign thyroid nodule and/or thyroid cancer. Before the FTC diagnosis, two patients had been treated with chemotherapy for pleuropulmonary blastoma (PPB) at two years of age (P2), or a primitive neuroectodermal tumor in the lung at five years of age (P5) (Table 2).
Discussion
We identified a genetic driver alteration in 80% of FTCs from patients younger than 20 years. Eight (53.3%) harbored a pathogenic DICER1 variant, including four with DICER1 syndrome. One (6.7%) had a germ line PTEN variant. A PAX8/PPARγ fusion was identified in one FTC (6.7%). Among five, multiple LOH with or without CNA, two (13.3%) had no coexisting variant, while the remaining three had coexisting DICER1 or PTEN variants mentioned above. Neither an RAS nor a TERT variant was detected in any FTC. Notably, all FTC tumors with a DICER1 variant had a variant at the metal binding sites within the RNase IIIb domain, and 87.5% had additional germ line or somatic variants across the DICER1 gene. Four patients (26.7% of 15 FTC patients; 100% in those diagnosed at age <10 years) were diagnosed with DICER1 syndrome with a pathogenic germ line DICER1 variant.
The DICER1 variant was predominantly detected in pediatric FTC patients in this study, whereas RAS variants and PAX8/PPARγ rearrangements were the most frequent genomic events in adult FTC patients (18,30). In addition, a germ line PTEN frameshift variant was found in one patient. A PAX8/PPARγ rearrangement was detected in one FTC, and no RAS or TERT variants were found; this suggests that the genomic architecture of pediatric FTCs is distinct from that of adult FTCs. A low frequency of RAS variants and/or PAX8/PPARγ fusions in pediatric FTCs was reported in two previous studies (19,20). However, the cited studies did not evaluate DICER1 status. Several pediatric studies on DICER1 variants have been conducted only in familial multinodular goiter patients and/or DTC in association with germ line DICER1 variants (31 –36). Recent NGS studies (17,37 –40) reported somatic DICER1 variants in 6.7% of FAs (n = 25) and 5.1% and 8% of FTCs (n = 30 and 39, respectively) (16,33), but not in PTCs (n = 125, 496, and 355, respectively) (17,39,40). However, these studies included patients of all ages, and only a few of the tumors were diagnosed in patients aged <20 years; thus, the findings seem to be representative of the frequency of adult tumors. Recently, pathogenic DICER1 variants have been identified in 3 (10%) of 30 adolescent-onset PTCs (41), suggesting a higher prevalence of DICER1 variants in the PTCs of young patients compared with adult patients. In the present study, although the number of patients is insufficient to draw any conclusions regarding the prevalence of genetic alterations in pediatric FTCs, we found that DICER1 variants played an important role as a major genetic alteration in pediatric FTCs.
Notably, our results suggest that the DICER1 syndrome should be considered in very young FTC patients, because all three children diagnosed with FTCs before the age of 10 years had DICER1 syndrome. Among them, a 9.3-year-old girl (P2), previously published by Shin et al. (42) and de Kock et al. (35), was diagnosed with PPB before FTC. However, a 2.4-year-old girl and 8.9-year-old boy initially presented only with FTCs but were eventually diagnosed with DICER1 syndrome. Regarding the DICER1 syndrome, recommendations for identifying at-risk individuals and surveillance strategies were recently published, with further updates to be expected (43). Because pathogenic germ line DICER1 variants are involved in >70% of PPB cases (44), genetic testing needs to be prioritized in all PPB patients (43). Based on our results, we suggest somatic DICER1 testing in children diagnosed with FTC, particularly if the child has a family history of multinodular goiter and/or thyroidectomy and/or history or coexisting history of an embryonal tumor. Two (P3 and P5) of our four patients with DICER1 syndrome in association with NH, FA, or FTC presented with multiple nodules at different stages, whereas nontoxic multinodular goiter was more frequently detected in elderly patients and the prevalence increased with age (45). Pediatric cases presenting with multinodular goiter should therefore be assessed for possible tumor development associated with DICER1 cancer predisposition syndrome.
Biological and molecular mechanisms explaining tumorigenesis in DICER1 cancer predisposition syndrome have been elusive. In addition to conferring increased risks of PPB and ovarian sex cord-stromal tumors, individuals with DICER1 syndrome may also develop lung cysts, cystic nephroma, renal sarcoma and Wilms tumor, genitourinary embryonal rhabdomyosarcoma, or embryonal brain tumors (43). Individuals with DICER1 syndrome typically show both truncating germ line DICER1 and second-hit hotspot missense variants. The DICER1 endonuclease is involved in microRNA (miRNA) production (31,46,47), and specific changes in DICER1 amino acids (p.E1705, p.D1709, p.G1809, p.D1810, and p.E1813) in the RNase IIIb domain result in defective cleavage of the hairpin loop structure of pre-miRNAs and a reduction of mature 5p miRNAs (36,48). Notably, all DICER1-positive tumors analyzed in this study (eight FTCs and six FAs) had one or two hotspot missense variants at metal binding glutamate or aspartate residues (49): p.D1709 in two FTCs and p.E1813 in six; p.E1705 in two FTCs and p.D1709 in one; and p.D1810 in one FA and p.E1813 in two (Fig. 1). In studies using murine Dicer knockout fibroblasts, inactivation of the RNase IIIb domain caused by the p.D1709A variant resulted in the elimination of 5p miRNAs, including let-7 (50). Although no specific miRNA or miRNA family has been identified as being involved in tumorigenesis, DICER1 mRNA expression showed an inverse relationship with let-7 and miR-345 levels (31,51), where let-7 is known to be necessary for thyroid follicular cell differentiation (52). All three human RAS genes contain multiple putative let-7 complementary sites in the 3′ untranslated regions, allowing let-7 to regulate RAS expression (53). Loss of the let-7 family can result in constitutive overexpression of the RAS oncogene and tumorigenesis (53,54).
To our knowledge, the c.5465A>G (p.D1822G) variant has not been reported previously; however, the other DICER1 missense variants identified in our study have been reported in thyroid lesions (32,36). We believe the novel variant (c.5465A>G, p.D1822G) is pathogenic because it has not been reported in a population database, including the 1000 Genomes Project and Exome Aggregation Consortium (55,56), and the variant was predicted to be deleterious (Sorting Intolerant From Tolerant [SIFT] score of 0) and a probably damaging (Polymorphism Phenotyping-2 [PolyPhen-2] score of 1 (57,58). These findings are shown in Supplementary Table S6. Similarly, another missense variant at the same amino acid codon (c.5465A>T p.D1822V) was inferred to be pathogenic in a patient with PPB (46) based on its location in the highly conserved residues of the RNase IIIb domain and the SIFT and PolyPhen-2 predictions. Furthermore, the in silico analysis predicted that transcripts with c.5465A>G had a higher chance of exon 25 skipping than those with the wild-type allele (59). Wu et al. reported that c.5429A>G and c.5438A>G not only function as missense variants (p.D1810G and p.E1813G, respectively) but also promote exon 25 exclusion from the DICER1 transcript (60), and that two additional variants (c.5441C>T and c.5428G>T) involved in skipping exon 25 resulted in a DICER1 protein incapable of producing 5p and 3p miRNAs (61). Although we could not perform functional studies of this novel variant (c.5465A>G), these findings suggest that it may be pathogenic, either as a missense variant (p.D1822G) or through skipping of exon 25.
The pathogenesis associated with progression of a normal thyroid to hyperplasia, and then to malignant transformation, has not yet been fully established. In two of our cases presenting with multinodular goiter, different second-hit missense DICER1 variants were present in each nodule diagnosed with FA and FTC, but not in NH in the same patient. Multiple distinct somatic hotspot variants have been reported in different thyroid nodules, including NH and thyroid cancer, in the same individual (32,34); however, we observed RNase IIIb hotspot variants in FA and FTC, but not in NH samples. It may be that germ line DICER1 variants predispose to thyroid hyperplasia, and the subsequent second hit of somatic DICER1 RNase IIIb hotspot variants leads to nodule development (32). DICER1 plays a role in terminating transcription by releasing polymerase II at sites of collision between transcription and replication, thus maintaining genome stability (62). Germ line truncating variants of the DICER1 gene may contribute to an environment in which second-hit variants can occur via adverse effects on genome stability or repair systems, which may in turn induce tumorigenesis. P7 and P8 in this study, who had two somatic DICER1 variants, also had a combination of one truncating and one missense (p.E1813K) variant; the former truncating variant may have occurred earlier than the p.E1813K variant due to the higher allele frequency, which tends to support the hypothesis delineated above (44). In particular, second-hit missense variants in all DICER1-related FTCs or FAs, regardless of whether there is a coexisting germ line DICER1 variant, occurred within sequences encoding the RNase IIIb domain, and especially at metal binding sites; this is consistent with previous reports (32,33,35,36,41). Along with the specific role of the RNase IIIb domain in processing miRNA, which is different from the RNase IIIa domain, the preference for somatic variants at the RNase IIIb domain suggests its importance in tumorigenesis. However, analysis of genetic alterations alone cannot fully explain the difference between FA and FTC development, so further studies are needed to better understand this distinct pathogenesis. In patients with a history of embryonal tumors (P2 and P5), antecedent chemotherapy may be associated with the pathogenesis of DICER1-associated FTC because the mutagenic effects of alkylating agents can accelerate tumorigenesis and reduce the latency of DICER1-associated thyroid cancer development (34).
Two cases (P11 and P12) had multiple LOH with or without CNAs, which were considered their main genetic alterations in the absence of other coexisting driver alterations. For the three FTC samples (P13–15) that failed to construct NGS libraries, we could only perform Sanger sequencing for hotspot regions of DICER, RAS, and TERT genes. If these FTC samples could be evaluated by more comprehensive methods such as NGS that cover multiple genes, it might be possible to identify the genetic driver alterations. The high success rate with respect to finding genetic driver alterations in this study (12 of 15 pediatric FTCs; 80%) also suggests that the genetic characteristics and pathogeneses of pediatric and adult FTCs differ.
Despite a large tumor size (>4 cm) in one-third of our patients, and coexisting angioinvasion in others, all patients showed favorable prognoses with NED during a median follow-up of 8.1 years, which may be attributable to the predominance of miFTC over eaFTC and wiFTC. While some reported that pediatric FTC patients present at more advanced stages, with larger tumors and lower rates of disease-free survival than adult FTC patients (16), no patients died from FTC (16,63,64). We found that the pediatric patients with FTC included in this study have a favorable course, regardless of the DICER1 variant. While a decrease in DICER1 gene expression may be associated with aggressive features in patients with PTC (51), those with DICER1 syndrome-associated DTC, which is predominantly diagnosed at a young age, may constitute a low-risk subgroup with low propensity for metastasis (36).
This study is limited by analyzing only 15 patients seen at a single center. However, the studied tumors could be analyzed with comprehensive genetic sequencing. Moreover, the FTC samples in this study were serially collected at a single center over 35 years, which minimizes selection bias. The predominance of DICER1 variants in pediatric FTCs needs to be validated in further studies with larger sample sizes.
In conclusion, pediatric FTCs have distinct genomic alterations and pathogenic mechanisms from adults, particularly characterized by prevalent DICER1 variants. Early-onset FTC in pediatric patients should raise suspicion for the presence of DICER1-positive tumors and DICER1 syndrome, especially in cases with multiple nodules, a family history of multinodular goiter, and/or a history of embryonal tumor.
Footnotes
Acknowledgment
The authors especially thank Jeong Seon Lee for sample preparation and molecular analyses.
Author Disclosure Statement
No potential conflicts of interest were disclosed.
Funding Information
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Science, ICT & Future Planning (grant number: NRF-2016R1A2B4012417), and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI13C2148). This study was also supported by the Seoul National University Hospital Research Fund (grant number: 04-2015-0830).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6