
Abstract
Background:
Discovery-scale omics datasets relevant to thyroid receptors (TRs) and their physiological and synthetic bioactive small-molecule ligands allow for genome-wide interrogation of TR-regulated genes. These datasets have considerable collective value as a reference resource to allow researchers to routinely generate hypotheses addressing the mechanisms underlying the cell biology and physiology of TR signaling in normal and disease states.
Methods:
Here, we searched the Gene Expression Omnibus database to identify a population of publicly archived transcriptomic datasets involving genetic or pharmacological manipulation of either TR isoform in a mouse tissue or cell line. After initial quality control, samples were organized into contrasts (experiments), and transcript differential expression values and associated measures of significance were generated and committed to a consensome (for consensus omics) meta-analysis pipeline. To gain insight into tissue-selective functions of TRs, we generated liver- and central nervous system (CNS)-specific consensomes and identified evidence for genes that were selectively responsive to TR signaling in each organ.
Results:
The TR transcriptomic consensome ranks genes based on the frequency of their significant differential expression over the entire group of experiments. The TR consensome assigns elevated rankings both to known TR-regulated genes and to genes previously uncharacterized as TR-regulated, which shed mechanistic light on known cellular and physiological roles of TR signaling in different organs. We identify evidence for unreported genomic targets of TR signaling for which it exhibits strikingly distinct regulatory preferences in the liver and CNS. Moreover, the intersection of the TR consensome with consensomes for other cellular receptors sheds light on transcripts potentially mediating crosstalk between TRs and these other signaling paradigms.
Conclusions:
The mouse TR datasets and consensomes are freely available in the Signaling Pathways Project website for hypothesis generation, data validation, and modeling of novel mechanisms of TR regulation of gene expression. Our results demonstrate the insights into the mechanistic basis of thyroid hormone action that can arise from an ongoing commitment on the part of the research community to the deposition of discovery-scale datasets.
Introduction
Cellular signaling pathways involving the two thyroid receptor (TR) members of the nuclear receptor superfamily, thyroid receptor alpha (THRA) and thyroid receptor beta (THRB), and their physiological bioactive small-molecule (BSM) ligands triiodothyronine (T3), thyroxine (T4), and 3,5-diiodothyronine (T2), regulate myriad aspects of cellular and systemic human physiology and metabolism (1). Broadly speaking, two basic syndromes result from changes in TR signaling, namely, thyrotoxicosis—possibly due to hyperthyroidism—and hypothyroidism. TRs are known to effect tissue-specific regulation of gene expression through a variety of mechanisms involving modulation of T3 transport and bioavailability, binding of TRs to thyroid response elements (TREs) in the promoters of direct target genes, recruitment of coregulators, as well as integration, or crosstalk, with pathways involving numerous other cellular receptors (2).
Discovery-scale datasets in the field of TR signaling have documented global effects of a variety of TR genetic and pharmacological manipulations on messenger RNA (mRNA) abundance (transcriptomics), and to a lesser extent promoter occupancy (cistromics), protein–protein interaction (interactomics), and cellular levels of metabolic intermediates (metabolomics). Although re-use of such datasets subsequent to their original publication has widely accepted potential for affording insights into the mechanistic basis of TR physiology, the infrastructure supporting such re-use is underdeveloped. For example, due to the time and effort required for their deposition, and lack of enforcement of deposition by publishers, many omics-scale datasets are absent from public archives (3,4). Moreover, those datasets that are archived are not presented in a state that encourages routine access and data mining. As a consequence, the value of these datasets has to date been underleveraged, representing a lost opportunity for progress in the field of TR signaling.
Motivated by a desire to enhance access for the research community to omics-scale datasets in the field of nuclear receptor signaling, we previously developed the Nuclear Receptor Signaling Atlas Transcriptomine transcriptomic dataset query tool (5). More recently, we expanded this initiative to incorporate a broad range of receptors, signaling enzymes, and transcription factors in a knowledge base referred to as the Signaling Pathways Project (SPP) (6). A central component of the SPP is the consensome (for consensus omics) meta-analysis algorithm, which ranks transcripts according to a measure of their significant differential expression in transcriptomic experiments involving genetic or pharmacological manipulation of pathway nodes in a given family (6). Here, we applied consensome analysis to publicly deposited datasets involving genetic or pharmacological manipulation of TRs in a murine tissue or cultured cell line. The resulting ranked lists of transcripts, referred to as TR transcriptomic consensomes—for all organs, liver and central nervous system (CNS)—were then validated against existing knowledge in the thyroid research literature. Finally, we provide a number of examples of the use of the consensomes to generate hypotheses around genes with elevated rankings in the consensomes that are previously uncharacterized as TR-regulated, but that have potential mechanistic roles in known facets of TR biology.
Materials and Methods
Biocuration of datasets
We first searched the Gene Expression Omnibus (GEO) or ArrayExpress to identify datasets involving genetic or pharmacological manipulation of TRs in a murine biosample (native tissue or cultured cell line). From this initial collection of datasets, we next carried out a three-step quality control check as previously described (6), which left a total of 20 datasets (Supplementary Data, Section S1; Fig. 1A). Next, within each dataset record, samples were grouped into all possible biologically appropriate experimental contrasts (referred to from here on as experiments, e.g., Thra null vs. wild type, Thrb knockdown vs. control, T3 vs. vehicle) manipulation of either TR isoform. Differential expression values were calculated for each gene in each experiment by using the limma analysis package from Bioconductor as previously described (5,7); then, they were committed to the consensome analysis pipeline as previously described (6).
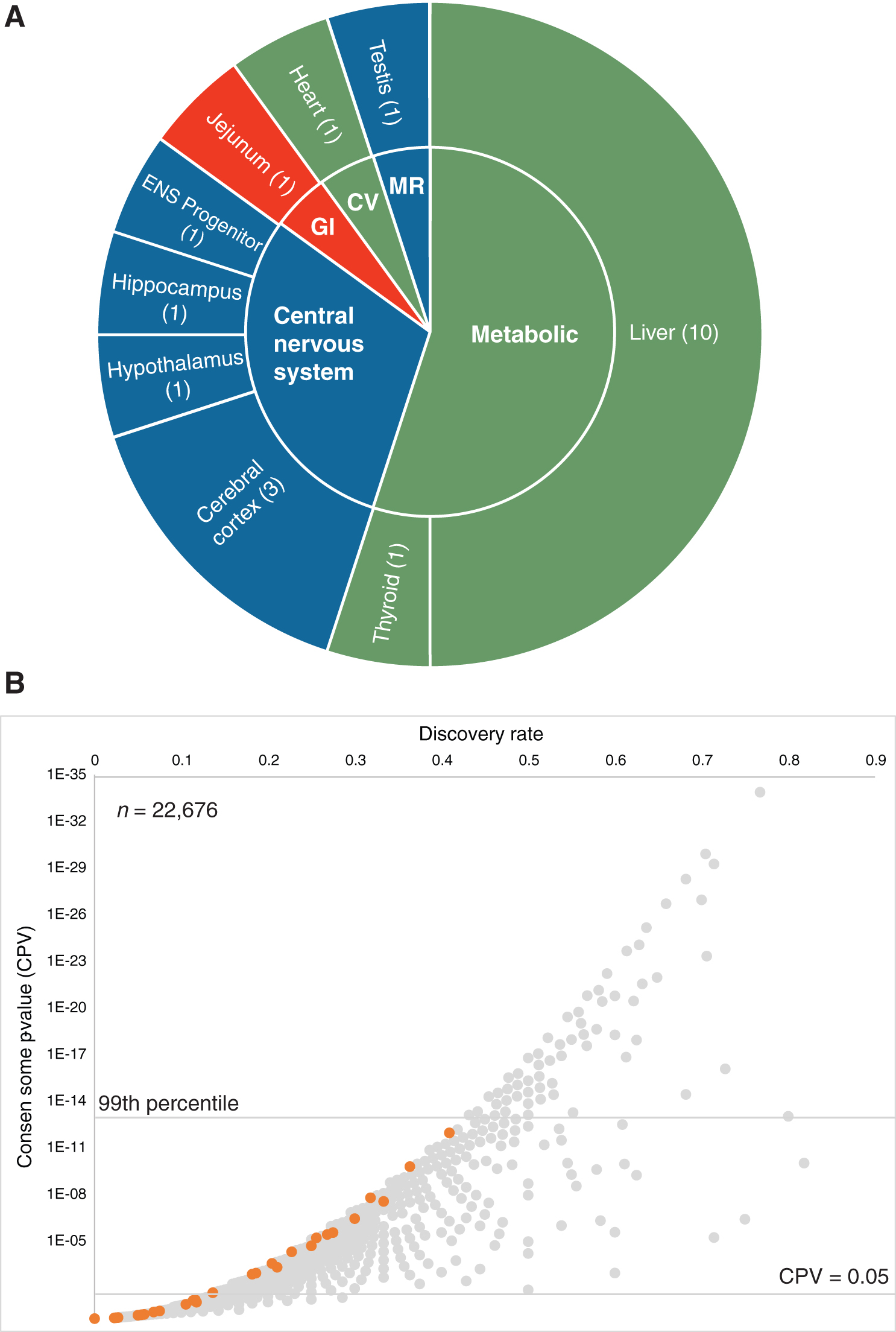
The mouse TR all organs transcriptomic consensome. (
Transcriptomic consensome analysis
Briefly, the consensome algorithm surveys each experiment across all datasets and ranks genes according to the frequency with which they are significantly differentially expressed. For each transcript, we counted the number of experiments where the significance for differential expression was ≤0.05, and then generated the binomial probability, referred to as the consensome p-value (CPV), of observing that many or more nominally significant experiments out of the number of experiments in which the transcript was assayed, given a true probability of 0.05. A more detailed description of the transcriptomic consensome algorithm is available (6). The consensomes were loaded into an Oracle 12c database and made available on the SPP user interface as previously described (6).
PANTHER over-representation analysis
Genes in the mouse TR liver and CNS transcriptomic 95th percentiles were submitted for PANTHER (version 14.1) Gene Ontology (GO) biological process over-representation analysis against the entire list of genes in the liver and CNS consensomes, respectively. Test Type was set to Fisher's Exact and Correction to “Calculate False Discovery Rate.”
Results
The mouse TR all organs transcriptomic consensome
A scatterplot depicting the mouse TR all organs transcriptomic consensome is shown in Figure 1B and the full consensome is in Supplementary Data, Section S2. To validate our approach, we first wished to determine the rankings assigned by our analysis to genes previously identified in the research literature as TR-regulated genes. As an objective measure, we retrieved a unique set of mouse orthologs of genes mapped to the GO terms “response to thyroid hormone” or “cellular response to thyroid hormone stimulus” that were present in the mouse TR all organs consensome (n = 43; Supplementary Data, Section S3). Of these, 60% (25/43) had CPVs ≤5E-02 (Fig. 1B, orange data points; Supplementary Data, Section S3). For the genes that did not have significant CPVs, we identified a number of considerations that might explain their nonsignificant rankings in the consensome, including genes incorrectly (Rdx, Six1) or ambivalently (Med1) annotated by GO; the use of nontranscriptional assays in the cited studies (Eif5a, Hspa9); and genes whose transcriptional regulation by TR may be specific to tissues and cell lines that were under-represented or absent from the datasets used to generate the consensome (e.g., Ghsr in the pituitary, Slx34a1 in the kidney). Supplementary Data Section S3 discusses these considerations in greater detail. Interestingly, of the 26 GO-annotated genes with significant CPVs, none was in the highest (99th) percentile of the TR all organs consensome (Fig. 1B).
We next wished to demonstrate the value of the consensomes in allowing researchers to routinely generate hypotheses addressing previously uncharacterized or underappreciated mechanisms underlying known aspects of TR physiology. To do this, we focused on the 90th percentile of the mouse TR all organs transcriptomic consensome (TC90), which equates approximately to a CPV of 4E-05 or lower (Supplementary Data, Section S2). We surveyed the mouse TR TC90 for transcripts with documented roles in a number of key facets of thyroid physiology and pathology, namely: lipid metabolism; carbohydrate metabolism; mitochondrial bioenergetics and biogenesis; neurogenesis and the CNS; and in blood clotting and wound repair, thyroid hormone bioavailability, and xenobiotic metabolism. Each of the tables given next identifies the gene symbol, namely, a brief summary of its function, its TR all organs consensome CPV and percentile ranking, and an indication of whether or not a transcript has been previously characterized as TR-regulated in the research literature. To illustrate the use of the consensomes to hypothesize previously uncharacterized mechanistic players in disorders of thyroid signaling, we identified human diseases associated with genes in the 95th percentile whose symptoms are manifest in organ systems with well-characterized links to such disorders (Supplementary Data, Section S4).
TR TC90 transcripts involved in lipid metabolism
The well-studied influence of thyroid hormones (THs) on lipid metabolism (8 –10) is evident in the variety of transcripts in the TR TC90, with roles in the synthesis, storage, uptake, and utilization of fatty acids, cholesterol, and other lipids (Table 1). The link between hypothyroidism and hypercholesterolemia is well established, and it is attributable in part to the fact that two genes known to be defective in familial hypercholesterolemia—Ldlr, encoding the low-density lipoprotein receptor (11), and Lpl, encoding lipoprotein lipase (12)—are characterized TR-regulated genes (Table 1). Exemplifying the ability of consensome analysis to highlight the role of previously uncharacterized TR-regulated transcripts in diseases of thyroid dysfunction, however, is the elevated ranking assigned to Ldlrap1, encoding an accessory factor with an essential role in the endocytosis of the LDLR-LDL-C complex in the liver (13). Another TR TC90 transcript whose regulation by TR is previously undocumented is Dhcr7 encoding 7-dehydrocholesterol reductase, which catalyzes the generation of cholesterol from 7-dehydrocholesterol using NADPH. DHCR7 is deficient in Smith–Lemli–Opitz syndrome, an inborn error of cholesterol metabolism with a broad range of neurological and developmental manifestations (14). Regulation of many lipid metabolism transcripts by TR is likely to be secondary to its transcriptional regulation of the gene encoding a nuclear receptor that is very closely associated with regulation of lipid uptake and storage, Pparg (15). For example, of the five members of the acyl-CoA thioesterase family represented in the 98th percentile of the TR TC90 (Acot1, Acot3, Acot4, Acot8, and Acot11), four are in the peroxisome proliferator-activated receptor (PPAR) murine transcriptomic consensome. Similarly, all three TR TC90 transcripts in the perilipin gene family, whose members play essential roles in the mobilization of lipids in adipose tissue (16), have elevated rankings in the PPAR transcriptomic consensome. On the other hand, of the three members of the hypoxia-inducible angiopoietin-like gene family in the TR TC90 (Angptl3, Angptl4, and Angptl8), only Angptl4 is in the PPAR TC90, suggesting that the others may be indirectly regulated by Hif1a (TR CPV 1.4E-05) or directly regulated by TRs.
Selected Murine Thyroid Receptor All Organs TC90 Transcripts Involved in the Regulation of Lipid Metabolism
CPV, consensome p-value; T2D, type 2 diabetes.
TR TC90 transcripts involved in carbohydrate metabolism
Signaling pathways involving TRs have well-documented roles in the regulation of hepatic carbohydrate metabolism (8). Broadly speaking, thyroid excess is characterized by increased generation and absorption of glucose, whereas hypothyroidism is associated with lower levels of glucose utilization in peripheral tissues (17). Consistent with this, numerous transcripts encoding enzymes and other factors with critical roles in carbohydrate storage, mobilization, and utilization are present in the TR TC90 (Table 2). Glucokinase (Gck) catalyzes the initial and limiting step in both glycogen synthesis and glycolysis (18) and is defective in a variety of diseases, including gestational diabetes, congenital hyperinsulinism, and type 2 diabetes (19). Phosphoenolpyruvate carboxykinase 1, the rate-limiting enzyme in gluconeogenesis and its eponymous rare genetic deficiency, is characterized by impaired gluconeogenesis and hypoglycemia (20). Other TR TC90 genes encoding enzymes in this category are muscle phosphofructokinase (Pfkm), encoding the rate-limiting enzyme in glycolysis; Pygl, encoding glycogen phosphorylase, which catalyzes the rate-limiting step in glycogenolysis; Gys2, encoding the rate-limiting step in glycogen synthesis; and G6pc, which encodes the enzyme catalyzing the hydrolysis of glucose-6-phosphate to glucose in gluconeogenesis and glycogenolysis. Reflecting the historical association between TR signaling and lysosomal function (21), variants in the human orthologs of Gys2 (22), Pfkm (23), Pygl (24), and G6pc (25) are associated with a variety of lysosomal disorders of glycogen storage. Other transcripts in the TR TC90 encode enzymes that do not themselves regulate reactions in carbohydrate metabolic pathways, but they have regulatory roles over those that do. Pyruvate dehydrogenase (PDH) is a multi-subunit enzyme that catalyzes the conversion of cytosolic pyruvate to mitochondrial acetyl-CoA, which enters the tricarboxylic acid cycle to ultimately generate adenosine triphosphate. The activity of the E1 subunit of PDH, which catalyzes the rate-limiting step in the reaction, is negatively regulated by phosphorylation by mitochondrial PDH kinase (PDK), comprising four isozymes with varying tissue expression patterns and phosphorylation site specificities (26). Genes encoding three of the four PDK isozymes (Pdk1, Pdk2, and Pdk4) are present in the 95th percentile of the TR transcriptomic consensome, potentially reflecting fine transcriptional control over PDK function by thyroid signaling. Although Pdk1 is known to be regulated by thyrotropin (TSH) (27), no previous evidence exists in the literature for regulation of PDK expression by TRs.
Selected Murine Thyroid Receptor All Organs TC90 Transcripts Involved in the Regulation of Carbohydrate Metabolism
TR TC90 transcripts involved in mitochondrial bioenergetics and biogenesis
A substantial body of evidence links thyroid signaling to mitochondrial biology, and in particular to positive regulation of energy homeostasis, including increased oxidative phosphorylation (28), browning of white fat (29), thermogenesis (17), and increased muscle turnover (30). Reflecting these associations, the TR TC90 assigns elevated rankings to a variety of enzymes, transporters, and other factors with well-documented roles in these processes (Table 3). An important thermogenic factor in the TR TC90 is Ppargc1b, which, similar to the related Ppargc1a, is selectively expressed in tissues with elevated oxidative capacity such as brown adipose tissue (31). Irisin is a thermogenic peptide whose exercise-induced secretion from skeletal muscle has been suggested to have beneficial effects on energy metabolism that extensively overlaps with those of TRs (32). The elevated ranking in the mouse TR consensome of the gene encoding the precursor of irisin, Fndc5, suggests that transcriptional induction of Fndc5 may contribute to enhancement by TR signaling of a variety of metabolic indices. Interestingly, irisin has been shown to inhibit TH production, suggesting the possibility of its contribution to the well-documented negative feedback loop regulating secretion of TH from the thyroid (33). Given that Fndc5 is known to be transcriptionally induced by Ppargc1a (32), it might be reasonably speculated that induction of Fndc5 by TR is secondary to induction of Ppargc1b, although transcripts downstream of Ppargc1a and Ppargc1b are not entirely overlapping (31). Members of the sirtuin family have attracted considerable recent attention as master regulators of lipid and carbohydrate metabolism and as potential determinants of lifespan (34). Sirt3 and Sirt5 encode mitochondrial members of the sirtuin family of deacetylases, which have a variety of prominent roles in cellular metabolism across multiple organ systems, including liver (35) and skeletal muscle (36,37). Of the two, Sirt5 is the better characterized, and it has been shown to be targeted by Ppargc1a (PGC-1α) and Prkab2 (AMPK) in the regulation of mitochondrial energy metabolism (38). Interestingly, in addition to the evidence within the TR TC90 for TR regulation of Sirt3 and Sirt5, T3 has been shown to regulate SIRT1 (39), suggesting that TH regulation of sirtuin levels may be a co-ordinated process involving a broad range of downstream transcripts.
Selected Murine Thyroid Receptor TC90 All Organs Transcripts Involved in Mitochondrial Bioenergetics and Biogenesis
ATP, adenosine triphosphate.
TR TC90 transcripts involved in neurogenesis and function of the neurosensory system
The role of signaling pathways involving TRs in the development and maintenance of the CNS is supported by abundant translational and clinical observations. In experimental animals, TH deficiency results in a range of CNS deficits, including impeded migration of cerebellar neurons, positional defects of Purkinje cells, abnormalities in cerebral cortex lamination, and deficient myelination (40,41). In humans, TH deficiency has a historically well-characterized association with cretinism and, among others, speech and hearing, locomotion, and behavioral indices (42 –44). Reflecting this association, a variety of genes encoding factors with important roles in neurogenesis and the maintenance of CNS integrity have prominent positions within the TR consensome (Table 4). Although a considerable volume of evidence connects TH signaling to myelination and neural connectivity (45,46), and the clinical association between disorders of thyroid signaling and Alzheimer's disease is well documented (47) the mechanisms underlying this association are not well known. Consistent with such a role is the elevated TR consensome rankings assigned to Bace1, encoding a beta-secretase implicated as a key player in the generation of the pathological amyloid plaque characteristic of the disease (48). Other transcripts encoding proteins with important roles in the maintenance of myelin integrity are Ndrg1, human variants of which are associated with Charcot–Marie–Tooth disease (49,50) and the mitochondrial aspartate-glutamate transporter Slc25a12, deletion of which in mice results in defects in myelination (51). Other TR TC90 transcripts with documented connections to neurodegenerative diseases include: Atxn1, human mutations in which have been associated with spinocerebellar ataxia, characterized by progressive loss of cerebellar neurons (52); Park7, defects in which are implicated in Parkinson's disease (53); and the prion protein encoded by Prnp, human variants of which run the whole gamut of neurodegenerative pathologies, including Creutzfeldt–Jakob disease, Huntington disease-like 1, and Kuru (54), and which has previously been shown to be regulated by thyroid-stimulating hormone (55). Finally, a substantial amount of research literature links the gap junction protein encoded by GJB2 to hearing loss and deafness (56), which has, in turn, historical links to hypothyroidism (57,58).
Selected Murine Thyroid Receptor All Organs TC90 Transcriptomic Transcripts Involved in Neurogenesis and Central Nervous System Function
CNS, central nervous system.
TR TC90 transcripts involved in other physiological facets of TR signaling
Blood coagulation and wound repair
A substantial body of clinical evidence links disorders of thyroid signaling to deficits in the hemostatic system, with hypothyroid patients at greater risk of bleeding, and hyperthyroid patients at increased risk of thrombosis (59). Consistent with this, numerous TC90 transcripts encode factors with pivotal roles in cell–cell and cell–cell matrix adhesion, wound healing, clotting, and fibrinolysis (Table 5). Components of the clotting cascade represented in the TC90 include genes encoding: the receptor for coagulation factor II, F2r; coagulation factor F7, deficiency in which causes congenital factor VII deficiency (60); and Apoh and Proc, which regulate inhibition of the coagulation cascade. The TR transcriptomic consensome also assigns an elevated ranking to Fn1, encoding the integrin ligand fibronectin, an important regulator of thrombosis (61) and to tissue transglutaminase (Tgm2), an integrin-binding adhesion coreceptor for fibronectin and a functional homolog of clotting Factor XIII (62). Consistent with its elevated ranking in the TR consensome, TGM2 has been previously shown to be downregulated in cells depleted of DIO1, encoding a deiodinase that catalyzes the production of active T3 from T4 (63).
Selected Murine Thyroid Receptor All Organs TC90 Transcriptomic Transcripts Involved in Other Aspects of Thyroid Physiology
TH bioavailability
An intricate system of endocrine feedback loops connecting the hypothalamus, pituitary and thyroid glands drives homeostatic regulation of TH levels in the blood (64). Although a number of components of this feedback infrastructure such as TR regulation of pituitary TSH subunit genes Cga (65), Tshb (66), and hypothalamic Trh (67) are well characterized in the research literature, these transcripts are not assigned elevated TR consensome rankings due to the absence (pituitary) or under-representation (hypothalamus) of available archived TR-relevant datasets. A number of other factors involved in pathways regulating TH availability are represented in the TR TC90 (Table 5). Some of these are characterized TR-regulated genes, such as Dio1 and Serpina7, which encode the major TH serum transport protein, and which are mutated in inherited T4-binding globulin deficiency (68). Uncharacterized TR-regulated genes in the TR TC90 involved in regulating T3 bioavailability include: Trhde, encoding a TSH-releasing hormone degrading peptidase; Ttr, encoding a T4 serum transporter that is mutated in transthyretin amyloidosis; and the iodide transporter genes Slc5a6 and Slc26a4, the latter of which is deficient in congenital hypothyroidism and Pendred syndrome (69,70).
Xenobiotic metabolism
Although the connection between disorders of thyroid function and the disruption of drug metabolism has been historically well characterized in the research literature (71,72), literature connecting thyroid signaling to regulation of xenobiotic metabolism is sparse. Despite this, the TR all organs TC90 is enriched for transcripts encoding xenobiotic detoxification enzymes in the glutathione-S-transferase (GST) (73), cytochrome P450 (CYP) (74), and carboxylesterase (CES) (75) enzyme families (Supplementary Data, Section S2), representative examples of which are shown in Table 6. Previous descriptions of TR regulation of GST family transcripts have been largely restricted to those encoding the alpha and mu subunits (76). Strikingly, of the 18 characterized members of the murine Gst gene family, 16 (88%) are in the TR family TC90, a nearly 10-fold enrichment over the expected number, and suggestive of a highly coordinated regulation of cellular Gst transcript levels by TR signaling (Supplementary Data, Section S2). Further, consistent with previous reports of regulation of CYP genes by TR signaling pathways (77) is the presence of 38 (37%) of the 102 known functional members of the mouse Cyp family in the TR TC90 (Supplementary Data, Section S2). Another family of enzymes whose role in drug metabolism has only relatively recently been appreciated is the CES, which catalyze the hydrolysis or transesterification of a variety of xenobiotics (78). Interestingly, more than 50% of the murine complement of 20 Ces genes are present in the TC90 (Supplementary Data, Section S2), an ∼4-5-fold enrichment over the expected frequency. Given that regulation of Ces genes xenobiotic nuclear receptors has been reported (79), and genes encoding the xenobiotic receptors Nr1i2/pregnane X receptor (PXR) (80) and Nr1i3/constitutive androstane receptor (CAR) (81) are assigned elevated rankings in the TR consensome (both CPV = 3.31E-07, 98th percentile), it may be that TR regulation of these and other detoxification genes is indirect through these receptors. Although PXR and CAR modulation of TH levels via transcriptional regulation of Dio1 has been previously documented (82,83), no literature report exists describing transcriptional regulation of either gene by TR signaling.
Most Highly Over-Represented Gene Ontology Biological Processes in the Thyroid Receptor Liver and Central Nervous System Transcriptomic Consensome 95th Percentile Gene Sets
FDR, false discovery rate; GO, Gene Ontology.
Insights into TR crosstalk with other receptors through consensome intersections
The focus to this point has been on the TR transcriptomic consensome itself and the insights it affords into potential mechanisms underlying TR physiology and pathology. However, signaling pathways do not function in isolation, and integration of signaling inputs, or crosstalk, is a well-characterized phenomenon in the field of cellular signaling (84,85). Plausible points of integration of TR signaling with other pathway nodes are readily apparent in the presence of transcripts encoding key cellular signaling pathway nodes and peptide BSMs in the TR consensome itself. Some of these have been previously characterized, such as regulation by TR signaling of genes encoding the transcription factors Bcl3 (CPV = 4.55E-25) (86), Hif1a (CPV = 2.8E-04)—a potential mechanism underlying TR regulation of Fn1 (87)—and the receptors Pparg (CPV = 2.43E-10) (88) and Ghr (CPV = 1.46E-07) (11207942). Others are uncharacterized in the research literature, such as transcriptional regulation of transcripts encoding the xenobiotic receptors Nr1i2 (CPV = 1.88E-09; encoding PXR) and Nr1i3 (CPV = 2.68E-15; encoding CAR), or of Hes1 (CPV = 1.65E-10), a transcription factor targeted by Notch receptor signaling and a potential point of integration between TR and Notch signaling (89).
Points of convergence between TRs and other cellular signaling pathways can also be hypothesized by identifying genes at the intersections of the TR consensome and those of other signaling nodes. The TR transcriptomic consensome is only one of numerous receptors, enzymes, and transcription factor transcriptomic and cistromic consensomes curated by the SPP (6). To illustrate the use of consensome transcript intersections in generating hypotheses addressing TR transcriptional crosstalk with other signaling modalities, we next identified genes present in the SPP transcriptomic consensome 99th percentiles (TC99s) for TR and three receptors with similarly pervasive metabolic and physiological roles, namely, glucocorticoid receptor (GR), insulin receptor (INSR), and leptin receptor (LEPR; Supplementary Data, Section S5). This list represents genes that consensome analysis predicts with high confidence to be regulated by TR and at least one of these other receptors. Later, for selected genes, we show how these consensome intersections can be used to generate hypotheses addressing the mechanistic basis for signaling crosstalk between these receptors.
TR: INSR crosstalk
Signaling by insulin and the INSR impacts myriad processes in anabolic metabolism of carbohydrates, lipids, and proteins across multiple organs and tissues (90). GTP cyclohydrolase I (encoded by Gch1) catalyzes the initial and rate-limiting reaction in the synthesis of the enzymatic cofactor, tetrahydrobiopterin (BH4), and variants of this gene have been implicated in dopa-responsive dystonia (91) and BH4 deficiency (92). Given the historical evidence linking both TR (93) and INSR (94) signaling to the regulation of BH4 levels, the presence of Gch1 in the TR (CPV = 1.1E-18) and human INSR (CPV = 4.1E-34) TC99s represents a possible point of confluence between these receptors in BH4 metabolism (Fig. 2, left panel). Similarly, a wealth of translational and clinical evidence links both TR (30,95) and INSR (96) signaling to diverse processes supporting skeletal muscle myogenesis and maintenance of muscle mass. Consistent with this is the presence in both the TR and INSR TC99s of Akirin1, a promyogenic factor shown to be involved in skeletal muscle regeneration (97) and myogenesis (98).
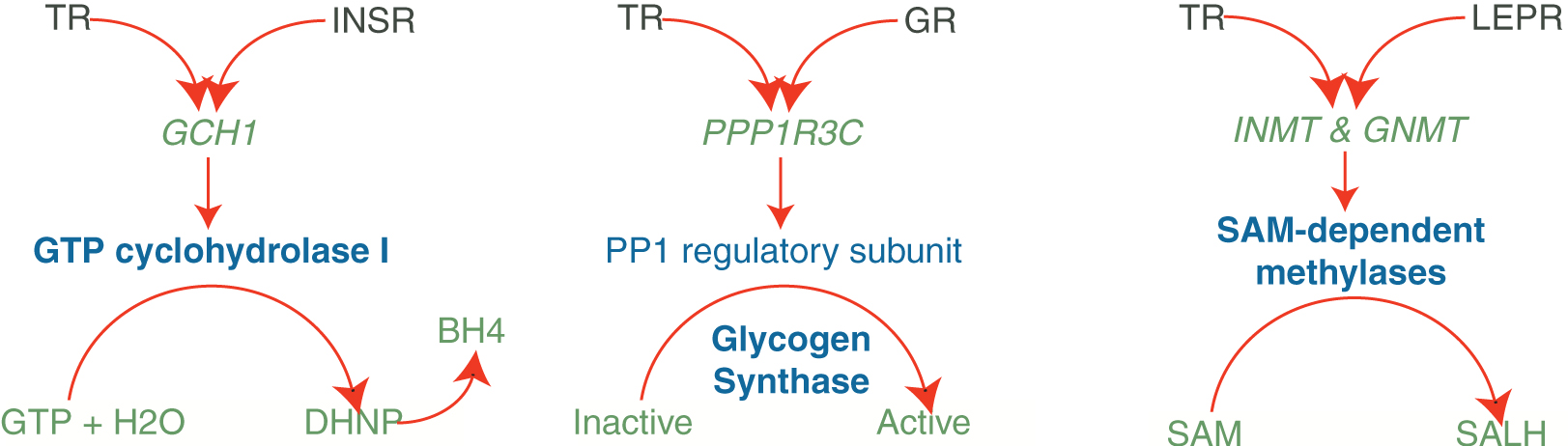
Consensome intersection analysis. Genes at the intersections of the TR, INSR, GR, and LEPR consensomes enable the generation of hypotheses around crosstalk between TR and these receptors in the regulation of metabolic pathways. See Supplementary Data Section S5 for a full list of genes. GR, glucocorticoid receptor; INSR, insulin receptor; LEPR, leptin receptor. Color images are available online.
TR: GR crosstalk
Reflecting its widespread anatomical distribution, the GR regulates gene expression in response to glucocorticoids in diverse neurological, physiological, metabolic, and immunological contexts (99). One such context is the regulation of macrophage differentiation and maturation (100), a process also shown to be regulated by TR signaling (101). A key transcription factor governing the expression of genes critical to macrophage differentiation is CEBPB (102); consensome evidence casting Cebp as a common TR and GR downstream regulated gene (Supplementary Data, Section S5) suggests regulation of its cellular levels as a plausible common mechanism for convergent TR and GR regulation of macrophage maturation. Another example of GR-TR crosstalk inferred from consensome analysis relates to carbohydrate metabolism, regulation of which by glucocorticoids (103) and TH (17,104) is supported by abundant literature evidence. The activity of glycogen synthase, a critical enzyme in maintaining the homeostatic balance between glycogenolysis and gluconeogenesis on the other, is regulated by protein phosphatase 1. The activity of this enzyme is closely regulated, in turn, by a regulatory subunit encoded by Ppp1r3c, which is present in the TC99s of both TR (CPV = 3.9E-20) and GR (CPV = 1.9E-15; Supplementary Data, Section S5), indicating possible convergent regulation of glycogen synthase activity by TR and GR signaling pathways (Fig. 2, middle panel).
LEPR crosstalk
Although identified initially as the cellular receptor for the adipokine leptin with a role in hypothalamic regulation of satiety (105), the functions of the LEPR are increasingly considered to extend much further. Although a growing volume of studies have characterized the regulation of TSH secretion by leptin (17,106), rather little is known regarding the integration of LEPR and TR-regulated signaling pathways. S-Adenosyl methionine (SAM) is a substrate in metabolic reactions involving transfer of methyl groups to numerous proteins, lipids, and secondary metabolites in signal transduction, protein repair, chromatin regulation, and gene silencing (107). Reactions involving SAM are catalyzed by enzymes collectively referred to as SAM-dependent methylases, two of which are indolethylamine N-methyltransferase, which plays an important role in the detoxification of selenium compounds, and glycine N-methyltransferase, an important player in methionine metabolism that is deficient in hypermethioninemia (108). The elevated rankings in the TR and LEPR consensomes of Inmt (CPVs: TR = 1.9E-11, LEPR = 3.32E-05) and Gnmt (CPVs: TR = 5E-15, LEPR = 3.32E-05; Supplementary Data, Section S5), which respectively encode these enzymes, are interesting in light of the previously established links between homocysteine and thyroid status on the one hand (109) and on the other hand, studies correlating serum levels of leptin and homocysteine in polycystic ovary syndrome (110) (Fig. 2, right panel).
Organ-specific TR consensome analysis indicates hat TR regulates distinct cell–cell adhesion gene networks in the brain and CNS
The mouse TR All-Organ transcriptomic consensome is intended as a rapid reference guide to evaluate evidence for transcriptional regulation of a specific gene by TR signaling, and the transcriptional responsiveness of that gene to TR signaling relative to other genes. Given that abundant evidence exists, however, to indicate that TR signaling exerts tissue-specific regulation of gene expression (111), we next wished to evaluate the extent to which such tissue-selective transcriptional effects could be modeled by using consensome analysis. Since they were the best represented in the datasets identified in our GEO survey, we generated TR consensomes for liver (Supplementary Data, Section S6) and CNS (Supplementary Data, Section S7) to identify evidence for genes that are selectively regulated by TR in one organ system over the other. For the purpose of this study, we used relatively stringent criteria, designating genes as selectively regulated if they were in the 99th percentile in one consensome and had a CPV of 0.5 or higher in the other. Applying these cut-offs, we identified genes selectively regulated in liver over CNS (Fig. 3A, Liver > CNS; Supplementary Data, Sections S6 and S7) and genes selectively regulated in CNS over liver (Fig. 3A, CNS > Liver; Supplementary Data, Sections S6 and S7). We also identified a third group of genes that had 99th percentile rankings in mouse TR consensomes for both organs (Fig. 3A, CNS ≈ Liver; Supplementary Data, Sections S6 and S7), indicating genes with close transcriptional regulatory relationships to TR in both systems. The relative positions of these three groups of genes are visualized in scatterplots (Fig. 3B) depicting the mouse TR liver (left) and CNS (right) transcriptomic consensomes. Genes predicted by this approach to be selectively regulated by TR in the liver include those encoding Abc and Slc family transporters, as well as enzymes catalyzing hepatic metabolic reactions, including galactokinase 1 (Galk1), Hagh, and St3gal3. Of the genes preferentially regulated in the CNS over the liver are Hap1 (112) and Daam2 (113) and they have been previously identified as TR-regulated; whereas Kcnj2, Galtn14, Zcchc12, and Foxg1 have no previously-established links to TR signaling (Fig. 3A; Supplementary Data, Sections S6 and S7).
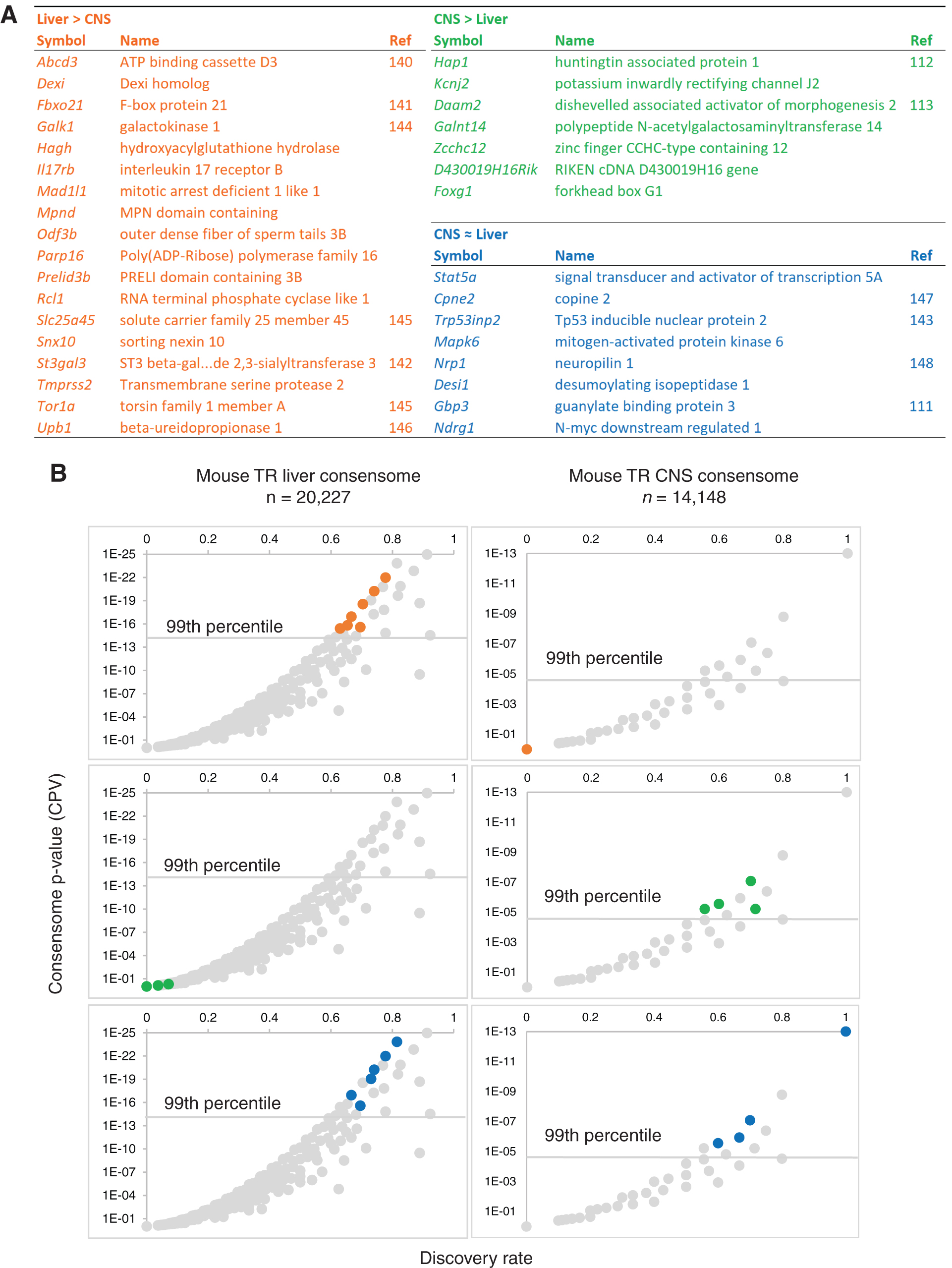
Liver- and CNS-specific TR transcriptomic consensomes highlight tissue-specific regulation of gene expression by TR signaling. (
For validation purposes, we evaluated the mouse TR CNS-specific consensome rankings of a set of 10 genes (Adamtsl4, Dbp, Fos, Hr, Kcna1, Klf9, Scd1, Stat5a, Trp53inp2, and Txnip) found by a previous meta-analysis (111) to (i) be differentially expressed in at least three TR CNS transcriptomic studies and (ii) possess a TR CNS binding site within 30 kb of the promoter start site (114). Of these nine putative direct TR targets, all but one were in the 89th percentile or higher of the mouse TR CNS consensome (CPV ≤1.1E-2; Supplementary Data, Section S7). The remaining gene, Fos (TR CNS CPV = 4E+1), has been previously shown to be responsive to T4 only in the presence of epidermal growth factor (115), which may help explain its relatively low ranking in the TR CNS consensome. To validate the TR liver-specific consensome, we examined the rankings assigned to a set of 10 genes (Car14, Chdh, Cyp2c29, Fabp2, Ldlr, Pnpla3, Pygl, Slc25a45, Srebf2, and Upb1) validated in a previous study (86) as direct TR transcriptional targets in the liver. Of these, all had significant rankings (CPV ≤1.8E-03) in the TR liver consensome that, with the exception of Fabp2, were in at least the 94th percentile (Supplementary Data, Section S6).
As an example of hypothesis generation using organ-specific TR consensomes, we next used the PANTHER resource (116) to carry out GO biological process over-representation analysis on genes in the 95th percentile of the liver and CNS consensomes (Supplementary Data, Sections S8 and S9), selected terms of which are shown in Table 6. The analysis reflects established roles for thyroid signaling in the regulation of specific aspects of liver metabolism (17) and CNS development (41), as well as provided evidence for relatively unexplored roles of TR signaling, such as in the modulation of noncoding RNA processes in the CNS. In addition, we noted a striking over-representation of genes in the GO cell adhesion category in the CNS (n = 84/723, false discovery rate (FDR) = 1.96E-09) relative to the liver (n = 39/983, FDR = 1) gene sets. Inspecting the TR CNS consensome 95th percentile, we noted that it contained an additional 13 genes that were related to GO-annotated cell adhesion genes and had published roles in that process not yet annotated by GO (Supplementary Data, Section S7). Although all 97 genes were present in the liver consensome, only 7% (7/97; Aqp4, Gas7, Nrp1, Nt5e, Rhob, Sned1, and Zfp36l1) were in the 95th percentile (Supplementary Data, Section S6). Interestingly, multiple members of distinct cell adhesion protein families are present in the liver and CNS TR consensome 95th percentile gene sets, the former containing members of the claudin (Cldn) and intracellular adhesion molecule (Icam) families, whereas members of the ephrin receptor (Eph), flibronectin leucine (Flrt), and protocadherin (Pcdh) families are exclusively represented in the TR CNS consensome 95th percentile (Supplementary Data, Sections S6 and S7). The contribution of cell adhesion pathways to synaptic plasticity (117), memory, and learning (118) is well documented, as is the connection of thyroid signaling to these processes (119,120). The research literature connecting thyroid signaling to cell adhesion comprises a set of focused paradigms such as TR regulation of neural cell adhesion molecule (Nrcam) (121). Our analysis adds to this body of knowledge by providing evidence that TR regulates an extensive network of cell–cell adhesion genes in the CNS that are largely distinct from those it regulates in the liver, and that many of these are previously unexplored as TR-regulated genes.
Discussion
Here, we have described the all organs, liver- and CNS-specific TR murine transcriptomic consensomes, which are intended as convenient reference resource that rank genes on the basis of their transcriptional responsiveness to genetic or pharmacological manipulation of TRs. The elevated ranking in the TR consensomes to genes previously characterized as TR-regulated establishes a level of trust in the user that the consensomes represents a reliable TR consensus transcriptional regulatory signature. In addition, the consensomes contain numerous examples of transcripts that, although previously uncharacterized as TR-regulated, possess functions that are appreciably aligned with known facets of TR cell biology and physiology. In addition to being valuable reference resources in their own right, the TR consensomes realize considerable added value through their incorporation into the wider SPP knowledgebase (6), facilitating an appreciation of other signaling nodes that converge on genes with elevated rankings in the TR consensomes.
The underdeveloped culture of dataset deposition by investigators and publishers in the field of cellular signaling has a variety of negative consequences for progress in research in the field. First, with the exception of well-funded laboratories supporting full-time informaticians with the expertise to computationally manipulate and mine these datasets, their rich store of information is effectively inaccessible for bench researchers. Second, rather than embarking on the challenging proposition of consulting these datasets to validate data or generate hypotheses, research funds are frequently spent either generating de novo datasets that already exist or on exploratory experiments based on partially developed hypotheses. The additional financial burden on already thinly stretched research budgets diverts funds away from experiments that advance knowledge in the field. With the availability of the TR consensome, insights into TR biology that were previously underappreciated due to the fragmented and challenging format of publicly archived datasets become more readily apparent and routinely accessible. For example, a powerful feature of the consensome user interface (6) is to facilitate the identification of regulatory relationships between TRs and genes encoding multiple members of a single gene family, most notably in the case of genes in the Ces, Cyp, and Gst family of xenobiotic metabolism enzymes (Supplementary Data, Section S2).
The current versions of the TR consensomes have some limitations. Expression array and RNA-Seq platforms inform only on mRNA abundance, and not on, for example, effects of TR signaling pathways on protein abundance or post-translational modification. Other limitations relate to the nature of the available archived datasets. For example, although many human TR-relevant transcriptomic studies are reported in the research literature, the vast majority of available archived transcriptomic datasets involving TR manipulation have been generated by using the mouse. Although a number of TR-relevant archived human datasets have been biocurated, these are currently not present in quantities that are sufficient to generate a statistically meaningful human TR consensome. Second, unlike other nuclear receptors, the volume of transcriptomic TR datasets is unfortunately not matched by the volume of archived TR ChIP-Seq datasets. This limits the potential for inferring the mechanism of regulation of genes in the consensome, which could potentially occur via direct DNA binding of TRs to TREs in the vicinity of their target gene promoters, or indirectly through other transcription factors, such as T3 regulation of SULT2A1 via SF-1 (122), or by effects on mRNA stability (123). Third, there is the potential for misleadingly high CPV values (and therefore low rankings) to be assigned to genes that are selectively regulated by TR in organs or cell types not represented in the TR all organs, liver or CNS consensomes.
Important as they are, datasets involving manipulation of TRs represent only a subpopulation of omics datasets whose re-use has the potential to catalyze the progress of research in the wider thyroid field. Case control datasets in studies of papillary, follicular, and anaplastic thyroid cancer, for example, can shed light on transcriptional changes that accompany the initiation and progression of these diseases. Second, datasets involving manipulation of any signaling nodes in the hypothalamus, pituitary, and thyroid gland biosamples have the potential to illuminate the existence of previously undiscovered signaling pathways governing the synthesis and secretion of T4, T3 and their regulatory peptides in these organs. To illustrate this, analysis of the mouse adipose tissue consensome identified a previously completely unstudied gene, Mcrip2, as a transcriptional target of numerous adipose signaling nodes with a potential role in adaptive thermogenesis (6). In the event of future research funding, we will extend consensome analysis to these additional populations of transcriptomic datasets.
The consensome approach is ultimately limited by the quantity and diversity of available archived datasets. In an ideal world, a sufficient number of quality datasets would be archived to permit the generation of a range of organ- and even cell type-specific consensomes. Although the findable, accessible, interoperable, and re-usable principles articulate an ideal for dataset stewardship (124), many datasets in the field of cellular signaling are not currently aligned with these standards. An ideal outcome of the availability of the TR transcriptomic consensomes will be to convey to researchers the tangible benefits that can accrue from archiving of their datasets, and to encourage them to deposit future datasets in the appropriate repository so that they can be incorporated into future TR consensome versions. By investing the effort in recycling its omics-scale datasets through public deposition, the thyroid research community can realize valuable opportunities to re-use these datasets for hypothesis generation and data validation, reduce unnecessary research expenditures, and perform more focused exploratory experiments. Finally, as statistical models, the TR consensomes can only provide evidence to support research hypotheses, and a truly comprehensive validation of the consensomes will require detailed experimental validation by the TR research community. We invite feedback from the community and invite investigators to assist in our efforts by contributing previously unarchived datasets for inclusion in future versions of the SPP knowledge base.
Footnotes
Acknowledgments
The authors appreciate the effort invested by investigators who archived their transcriptomic datasets, without which the consensome analysis would not be possible.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported in part by a grant from the American Thyroid Association and by the National Institute of Diabetes, Digestive, and Kidney Diseases NURSA (DK097748) and dkNET (DK097771) U24 awards.
Supplementary Material
Supplementary Data, Section S1
Supplementary Data, Section S2
Supplementary Data, Section S3
Supplementary Data, Section S4
Supplementary Data, Section S5
Supplementary Data, Section S6
Supplementary Data, Section S7
Supplementary Data, Section S8
Supplementary Data, Section S9