
Abstract
Background:
Sporadic medullary thyroid carcinoma (sMTC) is an uncommon neoplasia arising from the calcitonin-producing parafollicular cells of the thyroid. Previous studies evaluated whether single nucleotide polymorphisms (SNPs) within RET (a pivotal proto-oncogene for this disease) are associated with the risk for developing sMTC, but the results are inconclusive.
Methods:
In this work, we evaluated the association of RET-SNPs c.74-126G>T (rs2565206), p.Gly691Ser (rs1799939, G>A), p.Leu769 = (rs1800861, G>T), p.Ser836 = (rs1800862, C>T), and p.Ser904 = (rs1800863, C>G) (listed in the order of their chromosomal location) with sMTC. This is one of the largest case
Results:
sNMTC patients showed similar genotype and allele frequencies compared with healthy controls. On the other hand, among sMTC patients, the T-allele of p.Leu769 = was less frequent (OR = 0.70 [CI 0.58–0.84], p = 1.9 × 10−4) and rare homozygotes TT showed an OR = 0.32 ([CI 0.17–0.60], p = 2.3 × 10−4). Moreover, a statistically significant excess of the haplotype 2 (characterized by the alleles T-G-G-C-C of the listed SNPs) was observed (p = 3.9 × 10−3). The SNPs were not associated with the expression of RET mRNA, that is, they did not exert an effect in cis as quantitative trait locus (cis-eQTL). However, a strong eQTL association was found for p.Leu769 = and the neighboring gene CSGALNACT2 (p = 9.3 × 10−50; effect-size = −0.65), whose function in cancer is unknown.
Conclusions:
This study shows that specific RET haplotypes, in particular haplotype 2 and the T-allele of p.Leu769 = , are associated with a reduced risk of sMTC in Italians.
Introduction
Medullary thyroid carcinoma (MTC) is a rare neoplasia representing only 5–7% of all thyroid carcinomas (1). The familiar form of MTC (fMTC) is part of the spectrum of the multiple endocrine neoplasia (MEN) type IIA or B syndromes, inherited as an autosomal dominant disorder. However, sporadic MTC (sMTC) is the most common form, accounting for almost 75% of all cases (2). Usually, the diagnosis occurs between the age of 40 and 60 years (1,3) and the overall survival at 10 years is 99%, 93%, 71%, and 21%, for patients with stages I, II, III, and IV, respectively (4).
Inherited germline mutations within the RET gene (rearranged during transfection) were found to cause fMTC related to MEN, which is consistent with the expression of RET in the thyroid para-follicular C cells and in other adult endocrine tissues (e.g., the adrenal medullary chromaffin cells). Moreover, studies focusing on the sporadic form showed a high prevalence of somatic mutations affecting RET, with rates ranging from 57.8% (5) to 61.5% (6). According to COSMIC (Catalogue Of Somatic Mutations In Cancer), the prevalence of RET-positive samples on 2397 tested samples is about 44%, HRAS is found somatically mutated only in 10% of the samples, BRAF shows a prevalence of 6%, and KRAS mutations are found in 3%. In summary, RET is, by far, the most frequently mutated gene at the somatic level and it is acknowledged as a pivotal proto-oncogene for MTC.
The RET gene encodes a tyrosine-kinase receptor for the glial cell-derived neurotrophic factor-family ligands (7) and, on ligand binding, it plays a role in regulating cell proliferation, growth, differentiation, migration, survival, and apoptosis (8,9). Somatic alterations of RET were described also in up to 20% (10) of tissues from papillary thyroid carcinoma (PTC), the most common type of sporadic nonmedullary thyroid carcinoma (sNMTC), originating from follicular cells. However, contrary to MTC, the mutations in PTC consist of a spectrum of chromosomal rearrangements with currently nine reported fusions, RET/PTC-1 to RET/PTC-9 (11 –15). Their frequencies are increased up to 87% (16) among patients who had past exposures to ionizing radiations, such as in children subjected to the fallout of the Chernobyl nuclear accident (2).
After mutation, RET becomes constitutively activated and it triggers the MAPK and PI3K-AKT signaling cascade, causing an uncontrolled proliferation of the thyrocytes (17 –20). Reasonably, several authors wondered whether single nucleotide polymorphisms (SNPs) within RET (now on RET-SNPs) could be associated with altered risks to develop thyroid carcinoma. In particular, c.74-126G>T (rs2565206), p.Gly691Ser (rs1799939), p.Leu769 = (rs1800861), p.Ser836=(rs1800862), and p.Ser904=(rs1800863) were investigated for their association with MTC (20) but the results were inconclusive.
Meta-analyses were also performed and, apparently, only c.74-126G>T and p.Gly691Ser=were associated with the risk of MTC (21,22), whereas negative results were reported for p.Leu769 = and p.Ser836=(23). However, most of the studies included in the meta-analyses were carried out in different geographic areas, with possible differences in the ethnicity. Moreover, because of the rarity of the disease, limited numbers of cases were genotyped. Further, some of the studies reported only allele frequencies and were missing information about the genotype frequencies in cases and controls. Thus, the role played by RET-SNPs, if any, has to be further evaluated and large studies are needed. Here, one of the largest case
Materials and Methods
Study population
A series of 585 sMTC patients (234 males and 351 females) were recruited at the Endocrine Unit of the University Hospital of Pisa, Italy. They were selected on the basis of a negative family history of thyroid diseases and lacking known RET germline mutations. Moreover, they were deemed to have a low risk of MEN syndromes or other malignancies according to clinical, laboratory, and family data. No loss of follow-up or study end-data was observed in our subjects. Cancer-related death occurred in a single patient. The patients' mean age at diagnosis was 51.0 ± 14.9 years, and the median follow-up was 84.0 months. All affected individuals were subjected to total thyroidectomy and central neck dissection following standard procedures. Lymphadenectomy of the lateral compartment(s) was performed during the first surgery if lymph node metastases were already diagnosed or detected during the intervention. The sNMTC was another large group of 1530 Italian volunteers, as described elsewhere (24). Briefly, the initial diagnosis of NMTC may be based on the clinical suspicion, although final diagnosis was confirmed histologically or cytologically.
Controls (CNT) were 990 workers (390 males and 600 females) of the Cisanello Hospital of Pisa. They were contacted during their routine follow-up by the Occupational Medicine Unit, and their blood was collected at that time. The eligibility criteria were the same for cases and controls, and they included a minimal age of 18 years. For controls, the exclusion criteria were: All volunteers should not have been affected by any malignancy, chronic inflammatory disease, or related diseases in the past, and not have thyroid nodules, when known. A group of 540 extra controls (EC) were also collected at Meyer Hospital of Florence (Italy). They included 178 females and 362 healthy male blood donor volunteers with an average age of 51 ± 7.9. According to the Helsinki declaration, both healthy and affected volunteers gave their written informed consent to participate in the study, and the study protocol was approved by the local Ethical Committee (Azienda Ospedaliero-Universitaria Pisana, Comitato Etico Area Vasta Nord-Ovest, CEAVNO). The sample series presented in this work comprises and extends a preliminary analysis carried out in 2004 on 106 sMTC and 106 controls (25).
DNA extraction and analysis
For both patients and controls, the amount of blood remaining after performing clinical routine analyses was used for DNA extraction according to a previously described protocol (26). Sequence analysis of exons 10, 11, 13, 14, 15, and 16 of the RET gene was performed for research purposes with PCR amplification followed by standard Sanger sequencing (27).
The genotyping was performed by personnel blinded to the case - rs2565206, assay id#: C___3204331_10 - rs1799939, assay id#: C___3204350_10 - rs1800861, assay id#: C___7566349_20 - rs1800862, assay id#: C___7566348_10 - rs1800863, assay id#: C___3204358_10.
List of the RET-SNPs Genotyped in This Study
SNP, single nucleotide polymorphism.
Two percent of DNA samples were repeated blindly as quality control of the genotyping. Moreover, as additional quality control, exonic SNPs of MTC patients could also be matched with the electropherograms obtained from the sequencing of RET, when available.
In silico analyses
Linkage disequilibrium (LD) analysis was performed for the selected SNPs to characterize the presence of other nearby polymorphisms with an r 2 > 0.8. This was performed by using Haploreg V4.1. This tool also provides information for the analysis of the noncoding genome. Candidate regulatory SNPs are displayed together with the annotation of their protein binding sites (from the Roadmap Epigenomics and ENCODE projects). The information also considers the conservation of the sequence among mammals and the effects on the regulatory motifs and gene expression (from eQTL studies). Then, to evaluate possible biological effects of p.Leu769 = (the only SNP positive in our study) and its cognate SNPs in LD, we performed a series of additional computational predictions. We examined the gene expression level, as quantitative trait loci (cis-eQTL), available for each SNP by using the GTEx Portal. Moreover, SNPs were evaluated for their effect on miRNA binding sites by using the following tools: PolymiRTS Database 3.0, RNAhybrid, and MicroSnipers. To assess putative effects on splice enhancers or silencers, Human Splicing Finder (HSF) was employed. Finally, to evaluate putative effects on polyadenylation signaling we employed Poly(A) Signal Miner.
Statistical analyses
The statistical analyses were performed at the genotype and haplotype level. Individual genotypes or reconstructed haplotypes were evaluated with a multivariate logistic regression analysis (MLRA). The model returns the odds ratio adjusted (ORadj) for covariates (sex and age) and their 95% confidence intervals [CI] with a statistical p-value of the association, as described elsewhere (24). The most likely mode of inheritance was evaluated by performing an extended MAX test [as proposed by Hothorn and Hothorn (28)] based on multiplicity-adjusted p-values for the Cochran–Armitage trend test of the dominant, additive, and recessive models. Individual haplotypes were reconstructed from unphased SNP genotype data of unrelated individuals by using the software FastPhase. It implements methods based on a cluster model for haplotypes, as described in Scheet and Stephens. Parameters of the model are first estimated with an expectation–maximization algorithm; then, haplotypes are inferred (29). Individuals with an incomplete set of genotypes were excluded for haplotype reconstruction, and rare haplotypes (<1%) were excluded from the statistical analysis.
Each genotype was evaluated by the chi-square test for the Hardy–Weinberg equilibrium (HWE) in controls. Given that multiple comparisons were performed, Bonferroni's correction was employed, and 0.05/4 = 0.0125 was the novel significance threshold. This threshold was adopted, because p.Gly691Ser and p.Ser904=were in strong LD with each other (r 2 = 0.94). When using data from 1000 Genomes, Europeans were considered as a unique population in spite of different geographical origins. Heterogeneity test and meta-analyses were performed by using the free software MIX 1.7, as previously reported (21).
Results
The characteristics of sMTC and CNT are summarized in Table 2. Healthy controls were frequency-matched with sMTC cases for sex and they were about 2 years younger. The sNMTC patients showed a higher proportion of females, and they were about 4 years younger than healthy controls. For quality insurance, repeated genotyping yielded >99.5% of reproducibility and the genotype frequencies followed the HWE, with a minimal p-value of 0.18 for p.Ser904=at the chi-square test.
Characteristics of Controls, Sporadic Medullary Thyroid Carcinoma, and Sporadic Nonmedullary Thyroid Carcinoma Patients
CNT, controls; SD, standard deviation; sMTC, sporadic medullary thyroid carcinoma; sNMTC, sporadic nonmedullary thyroid carcinoma.
As reported in Table 3, none of the analyzed SNPs was associated with the risk of sNMTC, as the genotype frequencies among this group of patients were similar to those measured in CNT. For sMTC, the T-allele of p.Leu769 = was associated with a reduced risk with statistical significance holding also after Bonferroni's correction (ORadj = 0.70 [CI 0.58–0.84], p = 1.6 × 10−4). For the same SNP, the MAX test showed that the additive model was the best fitting model, with heterozygotes being at intermediate risk (p = 1.9 × 10−4). According to the MLRA, heterozygotes had a reduced adjusted OR (ORadj = 0.78 [CI 0.62–0.98]), statistically significant at the nominal level of 0.04 (not satisfying the Bonferroni's threshold); rare homozygotes showed an ORadj = 0.32 ([CI 0.17–0.60], p = 2.3 × 10−4), holding the statistical significance also after Bonferroni's correction. The other RET-SNPs were not associated with the risk for sMTC.
Association Between Risk of Sporadic Medullary Thyroid Carcinoma or Sporadic Nonmedullary Thyroid Carcinoma and RET-SNPs' Genotypes
Statistically significant results are reported in bold.
OR were adjusted for covariates.
CI, confidence interval; OR, odds ratio.
To further evaluate the role of RET-SNPs in sMTC, the power of the study was increased by analyzing another group of 540 EC (Table 4) collected in a different center of the same region and matched, as group average, for age. The results are in agreement with the previous findings. Also, when comparing sMTC with this group of healthy controls, the heterozygotes for p.Leu769 = showed a reduced risk to develop sMTC (ORadj = 0.79 [CI 0.61–1.03]), which was not statistically significant; rare homozygotes showed an ORadj of 0.21 ([CI 0.11–0.41], p = 5.1 × 10−7), which remained highly statistically significant also after Bonferroni correction. Subsequently, the same analysis was performed by using the genotype data available from 503 Caucasians from 1000 Genomes. First, we observed that the allele frequencies reported in 1000 Genomes were very similar to those measured in our series of healthy controls. Moreover, the comparison between sMTC cases and controls from 1000 Genomes confirmed that p.Leu769 = was the only polymorphism associated with sMTC with an ORadj of 0.37 ([CI 0.19–0.75], p = 0.004), which remained statistically significant also after Bonferroni correction (Table 4). In particular, the ORadj was 0.33 ([CI 0.12–0.87], p = 0.02) when considering the Tuscan (TSI) group (the group geographically most proximal to our population study) as controls. Finally, because of the similar genotype frequencies in controls and sNMTC, an analysis was performed by comparing sMTC against all non-sMTC volunteers, that is, sNMTC+CNT+EC+TSI to obtain higher statistical power. Heterozygotes showed an ORadj = 0.77 ([CI 0.63–0.94], p = 0.012) and the rare homozygotes an ORadj = 0.29 ([CI 0.16–0.52], p = 1.40 × 10−5), both statistically significant also after Bonferroni correction. The genotypes were also evaluated as haplotypes and the profile of the seven most frequent ones is reported in Table 5 for CNT+EC, sNMTC, and sMTC. For sMTC, a statistically significant excess of haplotype 2 (p = 3.9 × 10−3) was observed when compared with healthy controls or with sNMTC patients (p < 10−4 and p = 1.7 × 10−3, respectively). The excess of haplotype 2 is counterbalanced by a reduced frequency of haplotype 4 (p < 10−4). If haplotype 3 is taken as reference in an MLRA, haplotype 4 is associated with a reduced risk of sMTC (ORadj = 0.70 [CI 0.53–0.93], p = 0.01), while for haplotype 2 the ORadj is 1.22 [CI 0.97–1.65]. The rare haplotypes 5 and 6 also showed reduced and increased frequencies, respectively, but their absolute numbers were too small to carry out robust statistics. To assess a putative functional role for RET-SNPs, we carried out several in silico analyses. We found that p.Leu769 = together with other 24 SNPs (r 2 > 0.8) constitutes an LD block spanning RET and its neighboring locus CSGALNACT2 (Fig. 1). Although all the SNPs located within and nearby the locus were not associated with the expression of RET at the mRNA level, the SNPs of this LD block were found to be strongly associated with the mRNA expression of CSGALNACT2 in the thyroid. The two SNPs most strongly associated as eQTL (in LD each other with r 2 = 0.87) were p.Leu769 = (with an eQTL p-value of 10−48; effect size = −0.65), and NM_018590.4:c.-673G>A (alias rs2505512; p = 9.3 × 10−50; effect size = −0.65) located 337 base pairs upstream of the 5′ side of CSGALNACT2. However, it should also be noted that the other SNPs analyzed in this study and not associated with the risk of MTC were found to be associated with the mRNA expression of CSGALNACT2 (p.Gly691Ser: p = 10−5; effect size = −0.25 and p.Ser904=: p = 1.1 × 10−5; effect size = −0.25). No other relevant information was obtained from in silico analyses regarding possible effects on splicing, polyadenylation signaling, or miRNA binding sites of the 25 SNPs belonging to the LD block of p.Leu769 = , with the exception of NM_020630.5:c.*166T>A (alias rs2075913). This SNP is in almost complete LD (r 2 = 0.99) with p.Leu769 = and resides within the 3′UTR in the middle of a highly conserved region (Fig. 2), with a GERP (Genomic Evolutionary Rate Profiling) score of 5.05 (and an average value of 4.79 in a 14 base window surrounding the SNP). One predictor (HSF) suggested that this genetic variation could lead to the “Creation of an Exonic Splicing Enhancer (ESE) site” but with “probably no impact on splicing” (details are not reported for brevity).
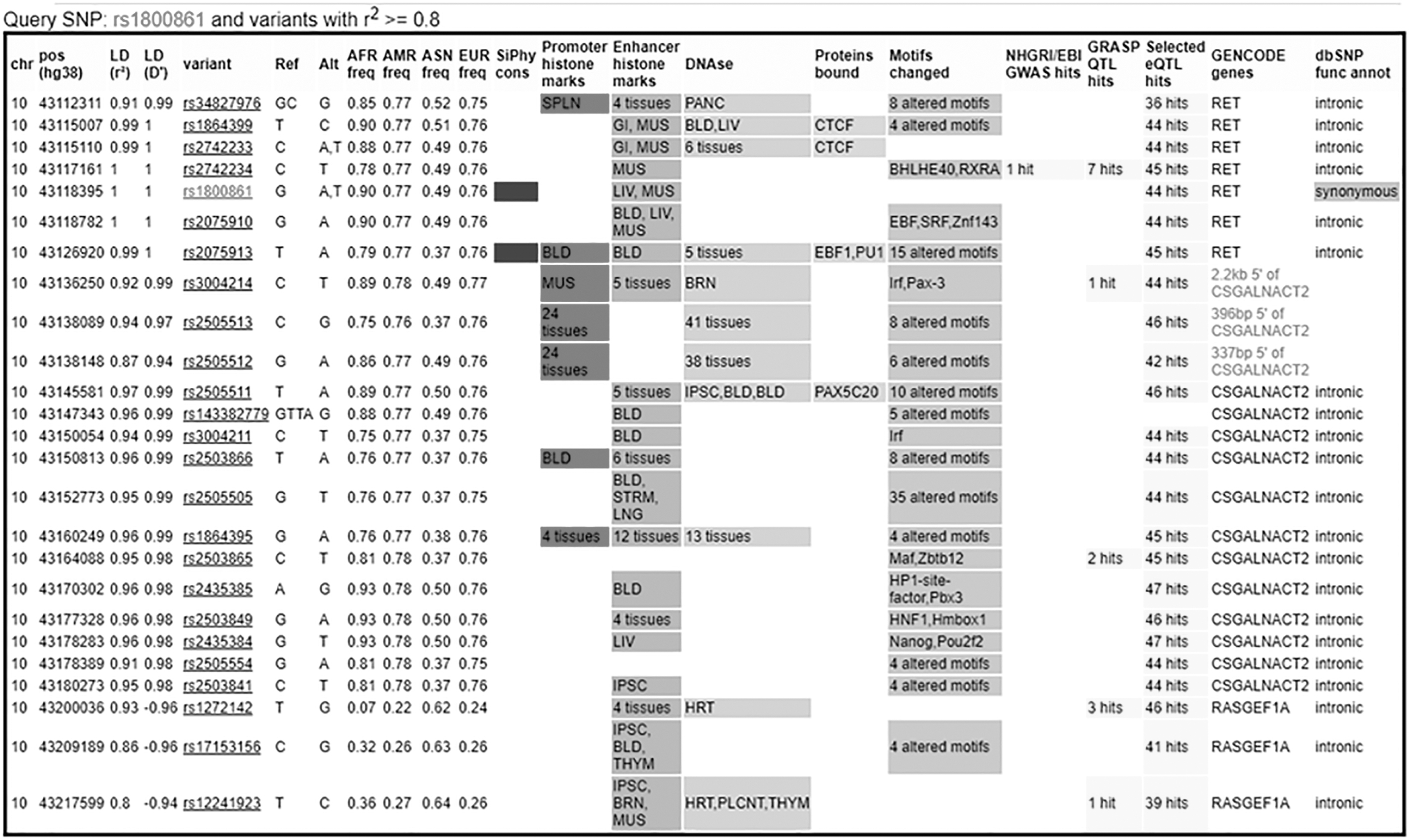
Snapshot of the output of Haploreg. p.Leu769 = (rs1800861) belongs to a block of LD together with other 24 SNPs. The analysis shows the possible or predicted effect of each genetic variation. This block of LD spans RET and its neighbor locus CSGALNACT2. LD, linkage disequilibrium; SNP, single nucleotide polymorphism.

Snapshot of the output from the UCSC Genome browser. c.3187 + 198T>A in tight LD with p.Leu769 = falls within a highly conserved region having a GERP score of 4.79 in a 14-base window. GERP, Genomic Evolutionary Rate Profiling.
Association Between Risk of Sporadic Medullary Thyroid Carcinoma and RET-SNPs' Genotypes, Considering Series of Healthy Controls from Different Sources
Statistically significant results are reported in bold.
OR were adjusted for covariates.
EC, extra controls.
Distribution of the Most Frequent Haplotypes Among Controls+Extra Controls, Sporadic Medullary Thyroid Carcinoma, and Sporadic Nonmedullary Thyroid Carcinoma
Haplotypes are shown with their allele composition in the order: c.74-126 (G>T), p.Gly691ser(G>A), p.Leu769 = (G>T), p.Ser836=(C>T), and p.Ser904 = (C>G).
Denotes a statistical significance.
Discussion
To ascertain whether RET-SNPs could be related to the risk of MTC, case
Here, we report that the sMTC group of patients is characterized by an excess in the frequency of haplotype 2, and a reduced prevalence of haplotype 4, as the consequence of the reduced prevalence of the p.Leu769 = T-allele. We found highly statistically significant ORs for G/T heterozygotes and uncommon T/T homozygotes with values of 0.78 and 0.32, respectively. In spite of the absence of an association reported for this SNP in a previous meta-analysis (23), it is unlikely that our results reflect chance for the following reasons. This study presents several strengths: (i) high statistical power: We collected, by far, the largest sample size ever analyzed in a single study on sMTC; (ii) lack of admixture of sporadic and familial forms: All patients were negative for a familial history of MTC and negative for mutations within RET after DNA sequencing; (iii) high quality of the reference population: All controls were healthy volunteers, showing genotype and allele frequencies that were remarkably similar to those found in other external databases (e.g., 1000 Genomes). Finally, the inclusion in the study of a second series of thyroid carcinoma patients (sNMTC) allowed controlling for possible occult selection biases that may occur when cases and controls have different compositions of distinct subpopulations (e.g., when patients chose to be hospitalized in geographically distant hospitals or were self-selected on the basis of their cost-sustainability for the hospitalization).
As p.Leu769 = is not expected to affect the protein, we evaluated whether the association could be ascribed to other causative variants in LD. We found a set of 25 SNPs in LD, with p.Leu769 = putatively affecting DNA motifs targeted by various transcription factors (Fig. 2). However, none of these SNPs was associated, as eQTL, with the expression of RET at the mRNA level. On the other hand, p.Leu769 = and NM_018590.4:c.-673G>A were found to be the two most strongly associated SNPs, as eQTL, with the expression in the thyroid of CSGALNACT2, a gene encoding for the chondroitin sulfate N-acetylgalactosaminyltransferase-2 and located just at the 3′ side of RET. This is a poorly studied type II transmembrane protein located in the Golgi apparatus. The literature reports only a possible involvement of this gene in pediatric high-grade glioma (30), and an altered expression at the mRNA level in multiple myeloma (31). Thus, biological evidence is lacking to understand the link between CSGALNACT2 regulation and risk of sMTC. Moreover, p.Gly691Ser and p.Ser904 = , both with an absent association with sMTC, were found to be associated with CSGALNACT2 mRNA expression, suggesting that this gene is probably not involved in sMTC susceptibility. As alternative, NM_020630.5:c*166T>A (alias rs2075913), which is in almost complete LD (r
2 = 0.99) with p.Leu769 = , could have biological consequences, as it is predicted to create a novel ESE by the HSF tool. However, the same tool indicated that an effect on splicing is unlikely. Finally, it could be speculated that, in the population considered here (i.e., southern and central Italians), there could be a founder mutation with intermediate penetrance conferring a risk for developing sMTC that is not sufficiently penetrant to result in obvious familial disease. The mutation could be in partial LD with specific SNPs, causing increased risks only for carriers of specific haplotypes (i.e., the haplotype 2). Because it is conceivable that different populations carry their own founder mutations, this hypothesis could also provide an explanation for geographically distinct patterns of risks, and for the low reproducibility among case
In summary, our study suggests that p.Leu769 = is a risk marker for sMTC, at least among central and southern Italians. As this locus is a robust candidate for MTC risk, future studies are strongly warranted. They should be aimed, in particular, at seeking the presence of low-frequency founder mutations hidden within the gene region and in LD with p.Leu769 = as well as at evaluating any association between p.Leu769 = alleles and the rates of maturation or translation of the RET transcripts.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was in part sustained by AIRC (Associazione Italiana Ricerca Cancro, investigator grant year 2008, Italy) and by the Istituto Toscano Tumori, grant system 2010, and Istituto Toscano Tumori (Italy) (Grant No. I56D15000010002).