
Abstract
Background:
Several mechanisms likely cooperate with the mitogen-activated protein (MAP)-kinase pathway to promote cancer progression in the thyroid. One putative pathway is NOTCH signaling, which is implicated in several other malignancies. In thyroid cancer, data regarding the role of the NOTCH pathway are insufficient and even contradictory.
Methods:
A BRAFV600E-driven papillary thyroid carcinoma (PTC) mouse model was subjected to NOTCH pathway genetic alterations, and the tumor burden was followed by ultrasound. Further analyses were performed on PTC cell lines or noncancerous cells transfected with NOTCHIC or BRAFV600E, which were then subjected to pharmacological treatment with MAP-kinase or NOTCH pathway inhibitors.
Results:
The presence of the BRAFV600E mutation coupled with overexpression of the NOTCH intracellular domain led to significantly bigger thyroid tumors in mice, to a more aggressive carcinoma, and decreased overall survival. Although more cystic, the tumors did not progress into anaplastic thyroid carcinomas. On the contrary, the deletion of RBP-jκ (a major cofactor involved in NOTCH signaling) did not alter the phenotype in mice. BRAFV600E-mutated PTC cell lines were resistant to pharmacological inhibition of the NOTCH pathway. Inhibition of MEK1/2 uncovered a predominant effect on Hes1/Hey1 transcription compared with NOTCH inhibition in BRAFV600E-mutated cell lines. Finally, γ-secretase activity and γ-secretase subunit transcription levels were dependent on ERK activation. Our findings suggest that MAP-kinase activity overrides the NOTCH pathway in the context of thyroid cancer.
Conclusions:
The interaction between the BRAF and NOTCH pathways demonstrates that the BRAFV600E mutation might bypass NOTCH and exert a strong positive effect on NOTCH downstream targets in thyroid carcinoma.
Introduction
Thyroid carcinoma is the most common endocrine cancer, ranked in ninth position among newly diagnosed cancers in 2018 worldwide (3.3% of all cancers) (1). Among the different subtypes, papillary thyroid carcinoma (PTC) is the most prevalent at 80% of all cases. PTCs can usually be well managed and the five-year survival rate is 95% (2). On the contrary, anaplastic thyroid cancer (ATC) is one of the most aggressive human cancers with a median survival of seven months, and despite its rarity, it accounts for almost half of the thyroid cancer-related deaths (3).
In PTC, BRAFV600E is the most common mutation occurring in about 45% of cases (4). This mutation mimics a phosphorylation in the activation segment of BRAF leading to a constitutive activation of the kinase (5). As a result, BRAFV600E-driven tumors exhibit high ERK phosphorylation, leading to unregulated cell proliferation (6). It has been shown in mice that knockin of BRAFV600E leads to the development of PTC (7). ATC can, at least in part, arise from well-differentiated carcinomas following the accumulation of additional mutations (8). This progression to ATC has been demonstrated in different mouse models, by combining BRAFV600E with other genetic alterations commonly found in human cancers, such as mutation of PIK3CA or deletion of PTEN (9).
Another player suspected to cooperate with BRAFV600E to promote anaplastic progression is NOTCH. NOTCH controls a highly evolutionarily conserved intercellular signaling pathway that regulates multiple cell differentiation processes during embryonic and adult life (10). NOTCH acts as a transmembrane receptor that can be activated by JAGGED or DELTA family ligands (11). These ligand/receptor interactions induce two subsequent cleavages that are performed, first by a disintegrin and metalloproteinase (ADAM) protease, and subsequently by a γ-secretase complex (12). These cleavages ultimately release the NOTCH intracellular domain (NOTCHIC), which then migrates into the nucleus. There, NOTCHIC interacts with RBP-jκ, a DNA binding transcription factor, and mastermind-like (MAML) coactivators (13) to induce transcription of target genes. Only a few targets are known: hairy and enhancer of split (HES), and hairy/enhancer-of-split related with YRPW motif protein (HEY). However, the effect of NOTCH might be wider than just inducing the expression of HES and HEY, since increasing evidence suggests cross talk between NOTCH and other major signaling pathways (14).
The crucial role of NOTCH in development has been intensively studied for more than 50 years, mainly in embryology (15,16). However, it has only recently been implicated in disease. A role for NOTCH in human cancer was first demonstrated in T cell acute lymphoblastic leukemia (17); NOTCHIC overexpression acts as an oncogenic driver. Later, the NOTCH pathway was identified as an oncogenic signal in other pathologies, including melanoma (18) and chronic lymphocytic leukemia (19). Conversely, NOTCH acts as a tumor suppressor in chronic myelomonocytic leukemia (20) and nonmelanoma skin cancer (21). It has therefore been termed a “double-edged sword,” as its role is expected to be cell specific.
The role of NOTCH in thyroid cancer is a subject of debate. Indeed, some studies claim that overactivation of the NOTCH pathway promotes thyroid carcinogenesis, metastasis, a worse prognosis, and progression to ATC (22,23). In contrast, other studies have shown that NOTCH signaling is downregulated in thyroid tumors and that NOTCH reactivation mediates growth suppression and the expression of differentiation markers. The latter would be suggestive for a potential role as tumor suppressor (24,25). Because of these controversial observations, we sought to further characterize the role of NOTCH in thyroid cancer.
Experimental Procedures
Mice
Braftm1Mmcm , Gt(ROSA)26Sortm1(Notch1)Dam , Rbpjtm1Hon , and Tg(Tg-cre/ERT2)1Kero (named, respectively, BRAFV600E, NOTCHIC, RBP-JκKO, and Thyro::CreERT2) mice are described elsewhere (26 –29). All mice were bred in a mixed FVB/C57BL6 background. Mice were detained in a 12/12-hour light/dark cycle room, and fed ad libitum. Induction of mutations and the tissue recovery protocol were also described previously (30).
Ultrasound imaging
Measurements were conducted each month using a VEVO 2100 High-Frequency Ultrasound system (VisualSonics) equipped with a 40 MHz transducer. Mice were anesthetized by inhaling a mixture of isoflurane (1.5%) and oxygen. The anesthetized animals were placed on a warmed examination table after neck shaving (Veet® cream). The tumors were imaged using a transverse view of the neck. Tumor burden was assessed by measurement of the surface of the biggest thyroid section picture observed on the anteroposterior axis, as described previously (7,9).
Histology
For immunohistochemistry, a mouse (K4006) or a rabbit DAKO kit (K4010) was used following the manufacturer's instructions. Hematoxylin/eosin, Masson's trichrome, TUNEL, and immunofluorescence staining was performed as described in a previous study (30). Primary antibodies were obtained from the listed commercial sources and diluted as follows: anti-GFP (D5.1) XP (1:200, 2956S; Cell Signaling), anti-RBP-Jκ (1:100, T6709; Institute of Immunology Co., Tokyo), anti-KI67 (1:300, ab16667; Abcam), anti-CK19 (1:300, TROMA-III; DSHB), anti-galectin-3 (1:300, ab53082; Abcam), and anti-vimentin (D21H3) XP (1:300, 5741S; Cell Signaling).
DNA isolation, Braf recombination, and Rbp-jκ deletion evaluation
DNA extractions were performed from ear punches by lysis of the tissue in NaOH 50 mM at 95°C for 30 minutes, and then buffered with Tris-HCl 1 M (pH 8). For mouse thyroid tissues, DNA was extracted by phenol/chloroform extraction using a QIAzol Kit (79306; Qiagen) following the manufacturer's instructions. Assessment of the recombination efficiency of Rbp-jκ and Braf in mouse tissues was determined by polymerase chain reaction (PCR) on DNA extracted from tumor sections or ear punches. The primers used in this study are listed in Supplementary Table 1. The PCR strategies and expected sizes of the amplicons for Rbp-jκ and Braf are described elsewhere (26,31).
RNA extraction and real-time PCR
RNA extraction and cDNA preparations were performed as described in a previous article (32). TaqMan or SYBR Green techniques were employed using the TaqMan gene expression Master Mix (Applied Biosystems); the products were analyzed on a ViiA™ 7 Real-Time PCR System (Applied Biosystems). Mouse Actb or human GAPDH was used as a housekeeping gene for normalization, and relative target gene expression was quantified by the 2−ΔΔCt method. Primers used for the TaqMan reactions were purchased from Applied Biosystems [Hes-1 (Hs00172878_m1); (4352341E)]. Primer sequences used for the SYBR Green reactions are listed in Supplementary Table 1.
Protein extraction and Western blotting
Protein extraction and Western blotting were performed as described in a previous study (33). Western blots were probed with anti-RBP-Jκ (1:1000, T6709; Institute of Immunology Co., Tokyo), anti-β-actin (1:1000, A2066; Sigma-Aldrich), anti-ERK1/2 (1:5000, 9107; Cell Signaling), anti-p-ERK1/2 (1:2000, 4370; Cell Signaling), anti-PAN-AKT (1:2000, 40D4; Cell Signaling), and anti-p-AKT-Ser473 (1:2000, 4060; Cell Signaling) antibodies. Secondary antibodies were anti-rabbit IR680 (926-68071; Li-Cor Bioscience) and anti-mouse IR800 (926-32210; Li-Cor Bioscience).
Cell lines and culture
The origin and genetic background of all cell lines used are compiled in Supplementary Table 2. HTh-104, KTC-1, and BCPAP cells were a gift from Prof. James Fagin (MSKCC, New York), and LNCaP cells were purchased from Sigma-Aldrich (89110211). All cell lines were cultured in RPMI medium (R8758; Sigma-Aldrich) except HEK-293 cells, which were cultured in Dulbecco's modified Eagle's medium (D5796; Sigma-Aldrich). Media of all cell lines were complemented with 10% fetal bovine serum (FBS) (10270; Gibco), 100 U/mL penicillin (P0781; Sigma-Aldrich), and 0.1 mg/mL streptomycin (P0781; Sigma-Aldrich). Media of HTh-104, BCPAP, LNCaP, and HEK-293 cells were additionally complemented with 1 × nonessential amino acids (5-13 K00; BioConcept). All cell lines were kept up to 40 passages or 6 months, whichever limit was reached first. Cell lines were validated using highly polymorphic short tandem repeat loci.
Transient transfections
HEK-293 cells were transfected at 60% confluency in six-well plates precoated with poly-
Drug manipulations
Cells were washed at 80% confluency with phosphate-buffered saline and starved 16 hours before treatment with their corresponding medium complemented with 0.5% FBS. Cells were treated with an MEK inhibitor (PD-0325901, 500 nM; AbMole M1763), a γ-secretase inhibitor (DAPT, 10 μM; AbMole M1746), or a combination of these inhibitors. A control condition was added by treating cells with 0.2% dimethyl sulfoxide (DMSO). Cells were harvested after 24 hours of treatment.
Membrane isolation and γ-secretase activity assay
The protocol used for the isolation of membranes is described elsewhere (37). Protein concentrations were quantified using the Pierce BCA protein assay kit (23225; Thermo Fisher), and 30 μg of protein was preincubated for 1 hour at 37°C with 50 μM of DAPT, 1 μM of PD-0325901, or no treatment as control. Proteins were then incubated with 20 μM of a fluorogenic γ-secretase substrate [NMA-GGVVIATVK(DNP)-DRDRDR-NH2] (565764; Merck), and the degree of substrate cleavage was measured over 200 minutes at 37°C by fluorescence in a microplate reader (GloMax Discover; Promega; excitation: 355 nm, emission: 440 nm). Fluorescence intensity was normalized to DMSO control.
Statistical analyses
Statistical analyses were performed using GraphPad software. The Mann–Whitney test was used to compare two groups, except for overall survival, where a Gehan-Breslow-Wilcoxon test was used. Real-time PCR results from PTC cell lines were analyzed by two-way ANOVA with Bonferroni post-test, and all other analyses were performed by one-way ANOVA with a Tukey post hoc multiple comparison. p-Values were under 0.05, 0.01, and 0.001, and were flagged with one to three stars, respectively.
Ethics approval
Mice were kept, treated, and euthanized according to Swiss federal guidelines. The experimental protocol was approved by the Vaud Cantonal Ethics Committee for Animal Experimentation (License No. VD2894).
Results
BRAFV600E and NOTCHIC overexpression worsens symptoms without inducing tumor progression
Our first aim was to assess the effect of NOTCH pathway overactivation in a BRAFV600E-driven PTC model. For this, we bred two cohorts of single BRAFV600E-mutated mice and double BRAFV600E/NOTCHIC-mutated mice, in parallel. For tumor burden analysis, we bred a cohort of 10 BRAFV600E and 9 BRAFV600E/NOTCHIC mice (Fig. 1A–C), and for aged-matched organ collection, we bred 10 BRAFV600E and 6 BRAFV600E/NOTCHIC mice (n = 6) (Fig. 1D, E). Expression of the mutated allele was induced by tamoxifen injection at the age of one month and the tumors were followed on a monthly basis per ultrasound imaging starting three months after induction. Both groups presented enlarged thyroids three months after injection (Fig. 1A). For comparison, a wild-type mouse thyroid cross section has a size of about 1 mm2 (data not shown). Histology of the thyroid of a wild-type mouse is presented in Supplementary Figure 1C and D. The average thyroid size in double-mutant mice was significantly higher compared with the single-mutant mice, starting at the first ultrasound measurement, and the difference increased over time (Fig. 1B). Mice were euthanized when presenting difficulties breathing and/or when the tumor burden reached 1 cm3, parameters that were defined as main “endpoint criteria” (Fig. 1C). Double-mutant mice reached the endpoint significantly earlier (median 179.5 days) than mice with a single BRAFV600E mutation (median 302 days). Double-mutant mice had significantly higher transcription of Notch-1 and Hey-1; Hes-1 transcription had only a slight tendency toward elevated expression (p = 0.0727) (Fig. 1D). Furthermore, GFP expression was detectable in the nuclei of double-mutant mice, confirming the successful recombination of the inducible NOTCHIC construct, since a nuclear tagged eGFP reporter gene had been cloned in tandem with the NotchIC downstream of an IRES (Fig. 1E).
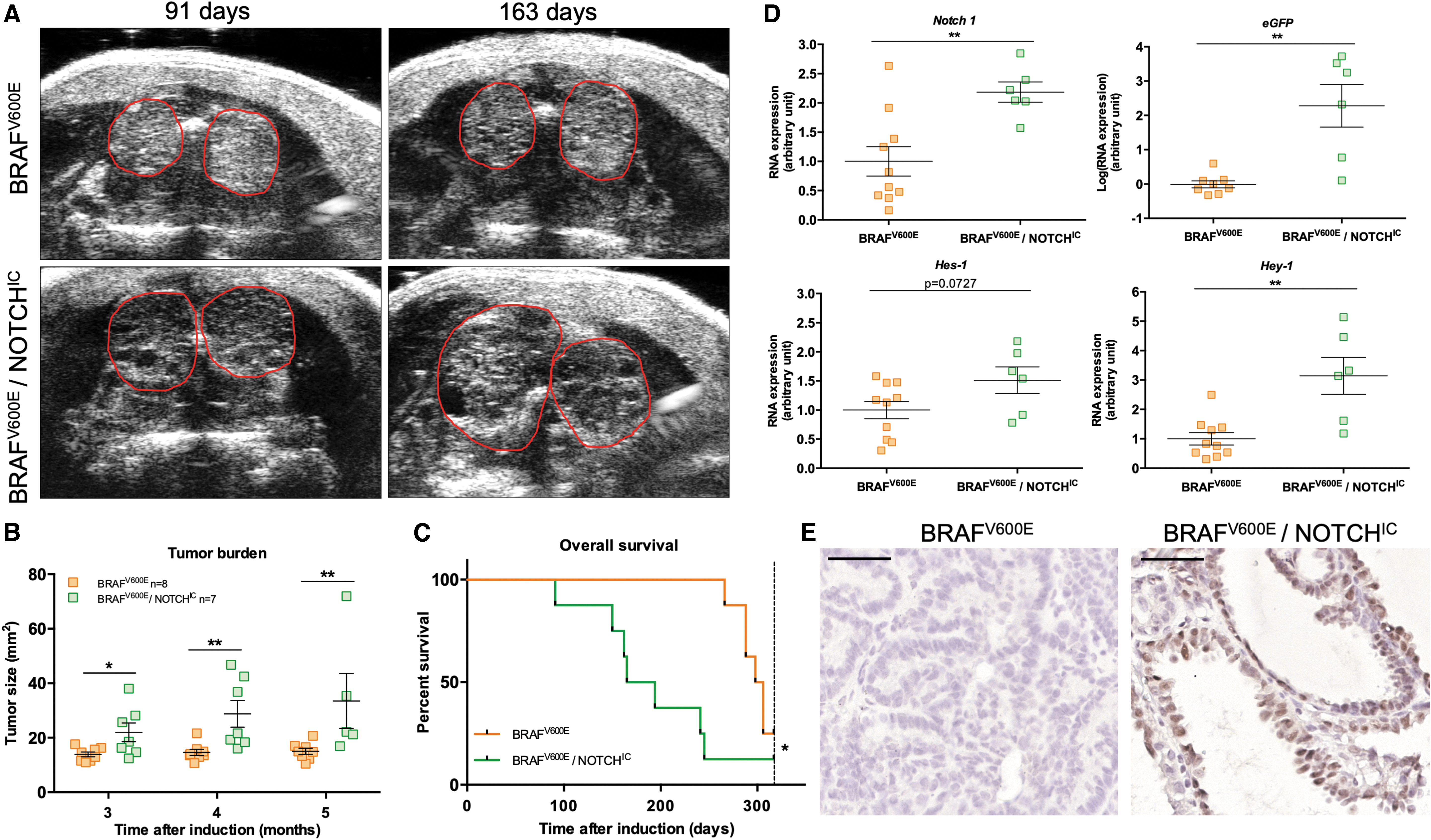
BRAFV600E and NOTCHIC overexpression led to larger tumors with a critical effect on survival in mice. (
Notch does not act as a tumor suppressor and is not necessary for PTC development
To further understand the role of the NOTCH pathway in PTC development, two cohorts of single BRAFV600E mutant mice and double-mutant mice for BRAFV600E/RBP-JκKO were bred in parallel. One cohort with BRAFV600E mice (n = 10) and BRAFV600E/RBP-JkKO mice (n = 9) was used for tumor burden analysis (Fig. 2A–C), and one cohort with BRAFV600E (n = 10) mice and BRAFV600E/RBP-JkKO mice (n = 6) was used for age-matched organ collection (Fig. 2D, E). Recombination was induced at the age of one month and the tumors were followed by monthly ultrasound measurements starting three months after induction (Fig. 2A). Both single-mutant and double mutant mice developed enlarged thyroids. The tumor burden was similar in both groups and it evolved comparably over time (Fig. 2B). This similarity in tumor size was paralleled by comparable overall survival (Fig. 2C). The median survival was 226 days for BRAFV600E mice versus 220 days for BRAFV600E/RBP-JκKO mice. To ensure that Rbp-jκ deletion was successful, we confirmed DNA recombination of the Rbp-j locus from DNA extracted from ear punches and thyroids in both groups (Fig. 2D). While all animals with an isolated BRAFV600E mutation showed a wild-type Rbp-jκ allele in ear punches and in the thyroid, double-mutant BRAFV600E/RBP-JκKO had a “floxed” allele in the ear punches and a recombined/truncated allele (Δ) in the thyroid samples, demonstrating the successful recombination/deletion of the gene. Mice carrying the deletion for Rbp-jκ had a lower overall level of RBP-Jκ protein (Fig. 2E). The model was further characterized by assessing RBP-Jκ protein expression in tissue sections. Sections from mice carrying the deletion had strongly reduced signals compared with single-mutated BRAFV600E mice in thyrocytes (Fig. 2F).

Notch activation is not necessary for the development of a PTC in mice. (
Mitogen-activated protein-kinase and NOTCH pathway overactivation does not promote ATC but rather leads to more aggressive PTC
Next, we performed immunostaining to define the subtype of the tumors (Fig. 3). Tumor cells from all groups were positive for PTC markers as demonstrated by positive galectin-3 and cytokeratin-19 signals, but were negative for the mesenchymal marker vimentin, which would have indicated epithelial to mesenchymal transition and ATC progression. Determination of the proliferation index using Ki67 revealed a significant difference between BRAFV600E (6.30% ± 1.19%) and BRAFV600E/RBP-JκKO mice (11.54% ± 1.69%) and no differences between BRAFV600E (7.07% ± 1.98%) and BRAFV600E/NOTCHIC mice (9.36% ± 3.18%) (Fig. 4A). TUNEL staining was then performed to monitor DNA fragmentation (Fig. 4B). Apoptotic bodies were quantified and no differences were found between any of the groups. Histological evaluation showed features of aggressive variants of PTC as tall cells and hobnail cells were exclusively found in the double-mutant BRAFV600E/NOTCHIC mice (Fig. 4C).
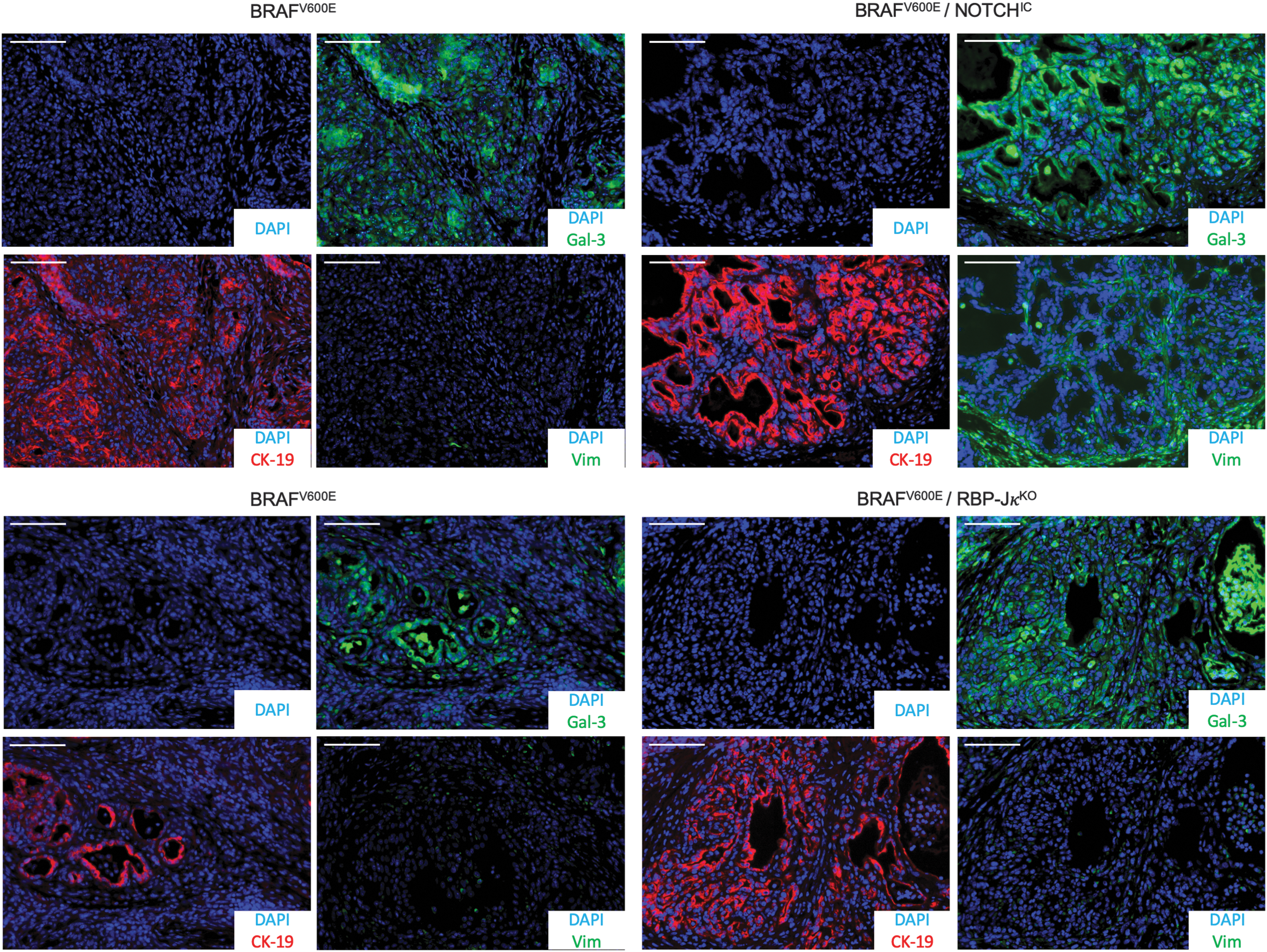
NOTCH pathway overactivation or downregulation in collaboration with BRAFV600E leads to PTC. Representative immunostaining pictures from BRAFV600E, BRAFV600E/NOTCHIC, and BRAFV600E/RBP-JκKO mouse thyroid tissues stained for GALECTIN-3, CYTOKERATIN-19, VIMENTIN, and DAPI. Scale bars: 100 μm.

BRAFV600E and NOTCHIC overexpression leads to more aggressive PTC. (
NOTCH pathway overactivation induces more cystic tumors
Histologically, BRAFV600E/NOTCHIC double-mutant mice tumors had more cysts (Supplementary Fig. 1A, B), which contributed to the overall enlargement of the tumors. It also suggests a remodeling role of NOTCH consistent with the fact that NOTCH activation can lead to collagen accumulation (38). However, Masson's trichrome staining did not show any differences in collagen content between single- and double-mutant mice tumors (Supplementary Fig. 2). Alternatively, the observed increase in tumor size could be associated with an upregulation of angiogenesis. Indeed, double-mutant mice had a higher CD31 signal (Supplementary Fig. 3A). NOTCH overactivation induces an upregulation of NOTCH ligands (39) in tumor cells, which could, in turn, activate the NOTCH pathway in endothelial cells and promote angiogenesis. Indeed, the tumors showed a slight elevation in Jag-1 and Dll-1 expression (Supplementary Fig. 3B), which could activate the NOTCH pathway and the proliferation of the endothelial cells present in close contact with the thyrocytes, thus promoting angiogenesis.
Elevated mitogen-activated protein-kinase pathway activity interacts with the NOTCH pathway by promoting expression of HES-1 and HEY-1
We elected to use three PTC cell lines with a BRAFV600E mutation (despite the fact that they bore other mutations, Supplementary Table 2) next used to investigate interactions between the mitogen-activated protein (MAP)-kinase and NOTCH pathways. Since all PTC cell lines known to us bare either BRAFV600E or RET/PTC, resulting in MAP-kinase elevation, we used LNCaP human prostate adenocarcinoma cells as a control, since they do not harbor a BRAFV600E mutation and low P-ERK. The mRNA expression levels of HES-1 and HEY-1 were quantified 24 hours after treatment with an MEK inhibitor, a γ-secretase inhibitor, or both (Fig. 5A). Interestingly, MEK inhibition strongly downregulated the transcription of both HES-1 and HEY-1 in PTC cell lines, but had no effect in LNCaP cells. Unlike MEK inhibition, γ-secretase inhibition had only a limited impact on HES-1 transcription and no effect on HEY-1 expression in PTC cell lines. By contrast, the transcription of both genes was strongly reduced in LNCaP cells after DAPT treatment. The combination therapy never had a greater effect than treatment with a single compound.
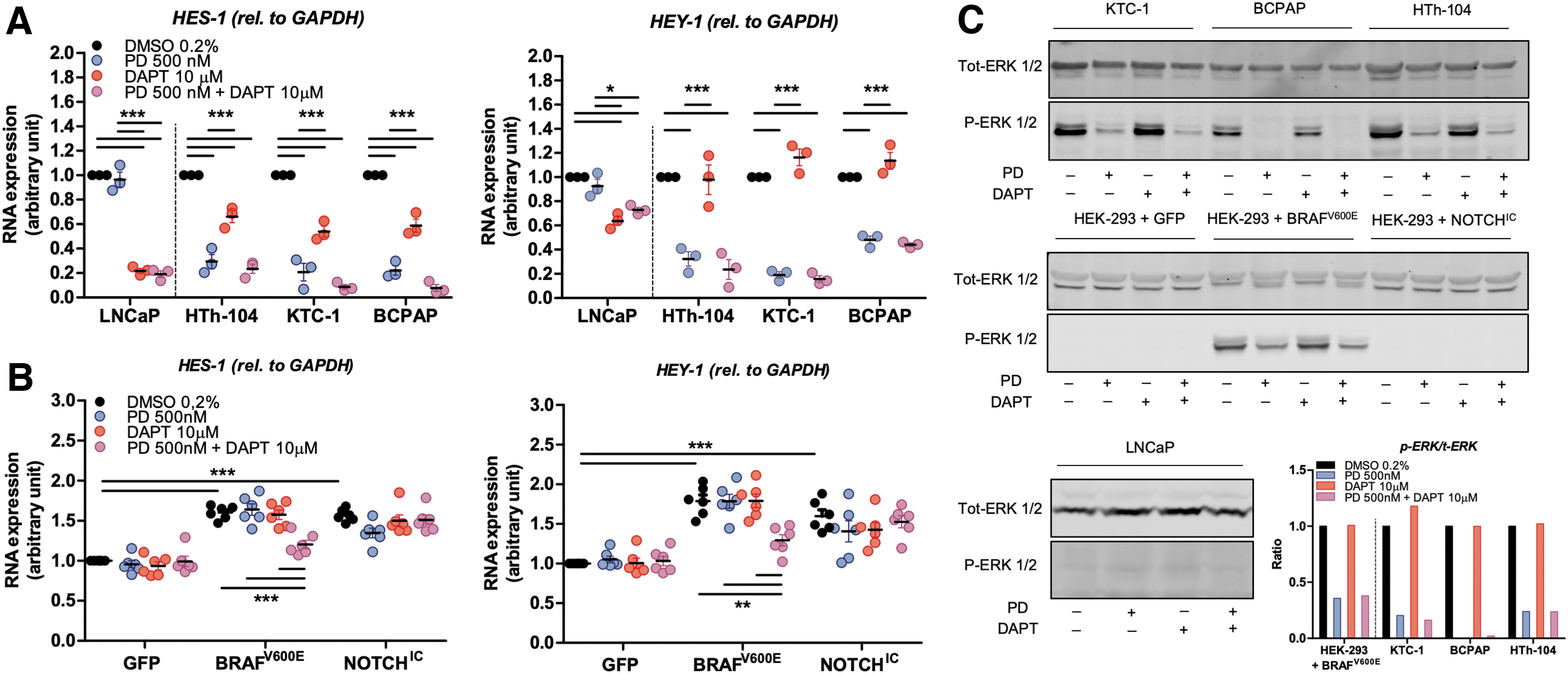
MAP-kinase pathway overactivation stimulates the expression of the NOTCH pathway's downstream target genes. (
To assess this interaction in a more dynamic model, HEK-293 cells were transfected with GFP (control), BRAFV600E (MAP-kinase overactivation), or NOTCHIC (NOTCH overactivation) plasmids (Fig. 5B). Both MAP-kinase and NOTCH overactivation led to a similar upregulation in HES-1 and HEY-1 transcription. This could only be rescued by pharmacologically blocking both pathways in cells transfected with BRAFV600E, which suggests interactions at different levels. It is important to note that the lack of an effect of DAPT on HES-1 and HEY-1 expression in NOTCHIC-transfected cells was expected because NOTCHIC is downstream of γ-secretase. The ratio of p-ERK over t-ERK was calculated to assess the efficacy of the MEK inhibitor treatment (Fig. 5C). While MEK inhibition highly reduced the ratio of p-ERK to t-ERK, in either single or in double treatments, DAPT had no effect on ERK phosphorylation.
MAP-kinase pathway overactivation also regulates NOTCH by targeting γ-secretase activity
To investigate any potential effects of the MAP-kinase pathway on the expression of γ-secretase subunits, we probed a thyroid tumor data set (GSE33630) from the Gene Expression Omnibus. We compared the expression level of all γ-secretase subunits between 49 PTC samples and 45 normal thyroid tissue samples. PSEN1 and APH1A subunits are significantly downregulated in PTC samples compared with normal thyroid tissue samples (Fig. 6A). Psen1 was also downregulated in BRAFV600E mice (Fig. 6B). Similar results were observed in HEK-293 cells transfected for 24 hours with pMKO.1 GFP, pBABEbleo-Flag-BRAFV600E, or 3XFlagNICD1 plasmids (Fig. 6C). Indeed, BRAFV600E transfection induced a significant decrease in PSEN1 expression, as did transfection with NOTCHIC. γ-Secretase activity in vitro has been determined in membrane protein extracts (Fig. 6D). LNCaP cells responded to DAPT treatment with a decreased γ-secretase activity compared with control cells treated with DMSO, while pharmacological blocking of the MAP-kinase did not affect γ-secretase activity in LNCaP cells. On the contrary, HTh-104 cells did not show a decrease in γ-secretase activity upon DAPT treatment, while PD-0325901 treatment did increase γ-secretase activity over time. BCPAP and HEK-293 cells did not respond to any treatments.
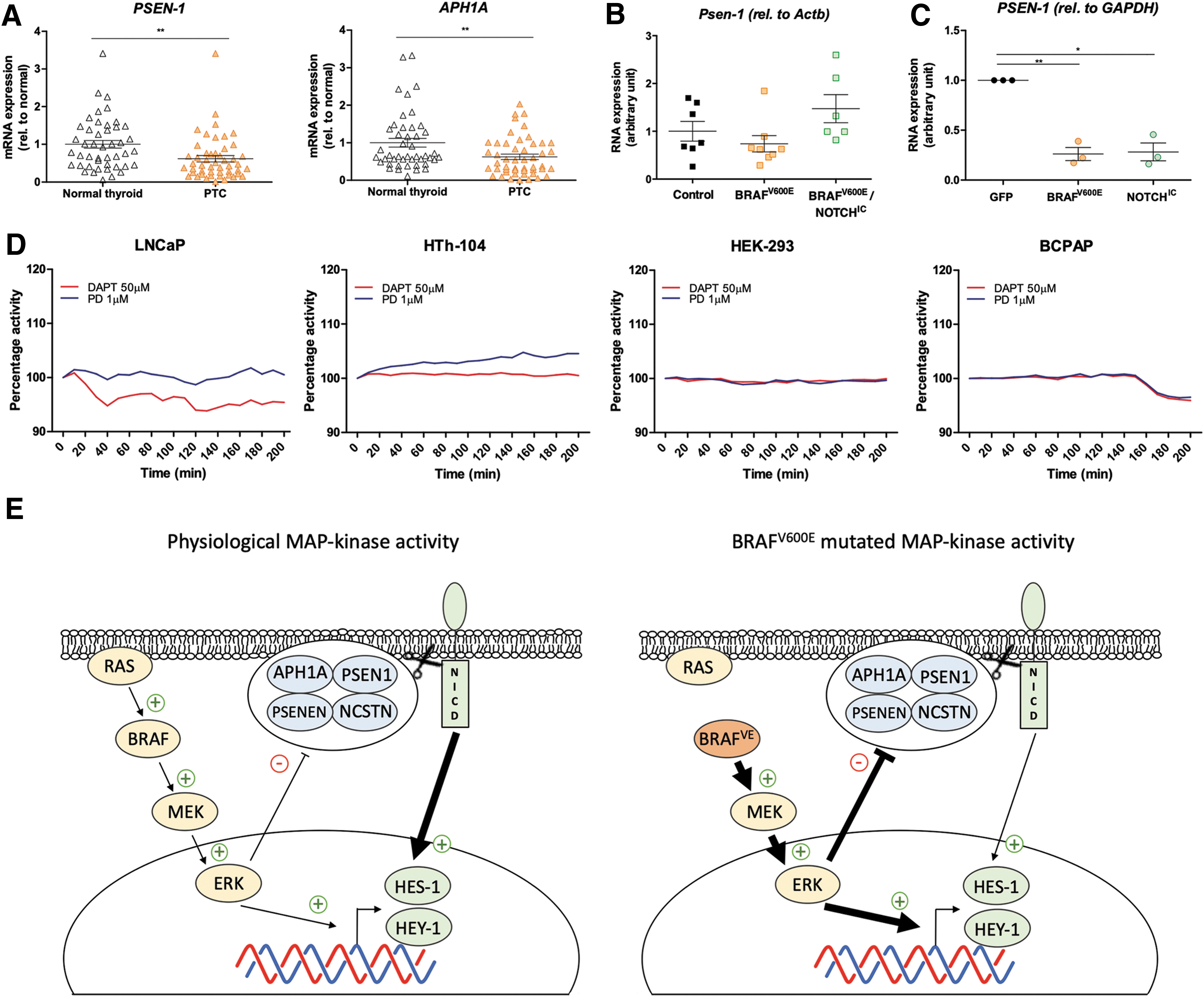
MAP-kinase pathway overactivity inhibits γ-secretase activity at the transcriptional and post-translational levels. (
Discussion
The role of the NOTCH pathway in thyroid cancer is unclear. In this report, we provide two new major insights into its role. First, we show that the presence of BRAFV600E combined with NOTCH overactivation (NOTCHIC conditional expression) in the mouse thyroid in vivo results in increased proliferation and larger tumors, and decreased survival (Fig. 1A–C), compared with mice harboring only BRAFV600E. The tumor burden was significantly increased from the third month onward. Double-mutant mice showed elevated transcription of NOTCH targets (Fig. 1D, E). This was further confirmed by the fact that the reporter gene eGFP, which had been cloned in tandem with NOTCHIC, was detected in the thyroid at the mRNA and protein level. This demonstrates that the strategy used was successful to activate both MAPK and NOTCH pathways. Interestingly, NOTCHIC alone could not induce the development of a goiter or a tumor (Supplementary Fig. 1C).
Double-mutant mice developed a more aggressive variant of PTC with tall cells (height at least twice their width) or a hobnail phenotype (Fig. 4C), both associated with shorter overall survival (Fig. 1C) (40,41). A clinical analysis investigating the relationship between clinicopathological factors and activation of the NOTCH pathway in 109 PTC cases showed that expression of activated NOTCH1 and HEY-1 was significantly related to histopathological variants and to the presence of BRAFV600E mutations (42). It would therefore be interesting to characterize the NOTCH pathway in tall cell and hobnail PTCs. The double-mutant mice did not develop tumors reminiscent of ATC, as determined by histological analysis and the absence of EMT; these findings suggest that NOTCH does not cooperate with BRAFV600E to promote a progression from PTC to ATC in mice, thereby contradicting some in vitro data where a potential role of NOTCH in the progression to ATC was suggested (23).
Our second major insight into the role of NOTCH in PTC concerns the fact that, NOTCH is considered a “double-edged sword” with both oncogenic and tumor suppressive roles depending on the context/tissue. We genetically deleted Rbp-jκ, a key player in the NOTCH pathway, to test whether NOTCH could work as a tumor suppressor in thyroid cancer. BRAFV600E mice combined with a knockout of RBP-Jκ developed tumors similarly to the BRAFV600E single mutants. In addition, mice with an isolated knockout of RBP-jκ did not develop a goiter or thyroid tumors (Supplementary Fig. 1D). Both results demonstrate that NOTCH does not have a major role as tumor suppressor in this thyroid cancer model, and indicate that the NOTCH pathway is not required for tumor initiation/formation. However, it has been shown that during development, total deletion of NOTCH or γ-secretase subunits results in a more profound phenotype than RBP-Jκ deletion (43); therefore, non-RBP-Jκ-dependent NOTCH effects on tumorigenesis cannot be formally excluded.
Increasing evidence suggests a possible interaction between the NOTCH and MAP-kinase pathways in neuroblastoma cells (44), as well as in pancreatic cancer cells (45). In thyroid cancer, it has been shown that the NOTCH pathway could be activated by the MAP-kinase pathway (46). However, the mechanism of this interaction remains poorly understood. To further investigate the interaction between these two pathways, we pharmacologically blocked the MAP-kinase pathway using an MEK inhibitor, the NOTCH pathway using a γ-secretase inhibitor, or both pathways in combination in vitro. These experiments were performed on BRAFV600E-positive PTC cell lines and on LNCaP cells (BRAFV600E-negative) as control. In the three PTC cell lines, the transcription of HES-1 and HEY-1 was strongly inhibited by MEK inhibition, but not affected by γ-secretase inhibition. In LNCaP cells, the expression of HES-1 and HEY-1 was marginally affected by MEK inhibition, but downregulated by γ-secretase inhibition (Fig. 5A, C). These results suggest that in the context of BRAFV600E mutant cells, HES-1 and HEY-1 are mainly driven by P-ERK and not by NOTCH. In addition, we observed that HES-1 expression is affected by γ-secretase inhibition in BRAFV600E-mutated cell lines, while HEY-1 expression is not. Through analysis of data in the Human Protein Atlas, we found that HES-1 mRNA is about 14 times more abundant than HEY-1 in the thyroid gland. This may be a reason why HES-1 expression is more affected than HEY-1 by γ-secretase inhibition in BRAFV600E-mutated cell lines.
To confirm these observations and to evaluate the interaction between the MAP-kinase and NOTCH pathways in a more dynamic setting, we transfected HEK-293 cells with plasmids encoding BRAFV600E, NOTCHIC, or GFP as control (Figs. 4 and 5B, C). BRAFV600E transfection induced HES-1 and HEY-1 upregulation, slightly more than NOTCHIC transfection did. Combined with the fact that PTC cell lines showed resistance to the inhibition by γ-secretase, this finding suggests that the MAP-kinase pathway overrides NOTCH signaling by acting on the downstream gene targets of NOTCH.
In vitro, the combination of both γ-secretase and MAP-kinase inhibition in BRAFV600E-transfected HEK-293 induced a decrease in HES-1 and HEY-1 transcription, while either drug alone had almost no effect. This suggests that in a BRAFV600E-driven cell, inhibiting MEK or the γ-secretase activity alone is not sufficient to induce a downregulation of the downstream gene targets of NOTCH, presumably due to possible feedback inhibition and cross talk. Based on these observations, we hypothesize that BRAFV600E expression induces elevated HES-1 and HEY-1 expression and inhibition of γ-secretase activity. When PD-0325901 is added, HES-1 and HEY-1 are no longer driven by the MAP-kinase pathway, and simultaneously, γ-secretase activity repression is abolished. The combination of these two effects resulted in NOTCH-driven positive regulation of HES-1 and HEY-1. Therefore, only the inhibition of both the NOTCH and MAP-kinase pathways can result in a reduction of HES-1 and HEY-1 transcription to almost basal levels (Fig. 5B).
Interestingly, the expression level of γ-secretase subunits consistently showed a downregulation of the PSEN1 subunit in vitro, in vivo, and in the clinical samples (Fig. 6A–C). APH1A seemed also to be downregulated in the patient samples, but not in BRAFV600E-HEK-293-transfected cells or in BRAFV600E mice (data not shown). PSEN1 is the catalytic component, and APH1A plays an important role for the proper assembly of the different subunits of the γ-secretase complex (47). Overall, our analysis suggests that besides the direct inhibition of MAP-kinase on γ-secretase activity, MAP-kinase overactivity also has a transcriptional inhibitory effect on at least PSEN-1 and possibly also APH1A.
It has been previously reported that ERK activation can reduce γ-secretase activity by inducing alterations in the phosphorylation of different subunits of the γ-secretase complex (37,48). In the present study, we observed this inhibition of activity in one PTC cell line (Fig. 6D) using a γ-secretase substrate that releases a fluorescent peptide upon cleavage. HTh-104 had increased fluorescence upon PD-0325901 treatment, which reveals an increase in γ-secretase activity potentially due to the inhibition of MAP-kinase activity. Interestingly, BCPAP, another PTC cell line, did not respond to any of the treatments. The variability observed between the cell lines indicates that the mechanism might be even more complex and depend on other mutations in the cells. However, it is interesting to see a lack of an effect on γ-secretase activity in BCPAP in response to the different treatments since HES-1 and HEY-1 gene expression was significantly downregulated when the MAP-kinase pathway was pharmacologically blocked in this cell line (Fig. 5A). These results also support a direct action of the MAP-kinase pathway on the transcription of the downstream gene targets of NOTCH, HES-1, and HEY-1. It is surprising that DAPT did not affect γ-secretase activity in HEK-293, suggesting that the NOTCH pathway is “inactive” in these cells. However, it is consistent with the data demonstrating no effect on the expression level of HES-1 and HEY-1 during DAPT treatment of GFP-transfected HEK-293 cells (Fig. 5B). This suggests that neither the MAP-kinase nor the NOTCH pathway is substantially active in nontransfected HEK-293 cells.
In conclusion, the data presented here provide new insights into the role of NOTCH in thyroid cancer. The findings demonstrate that MAP-kinase has a predominant effect on HES-1 and HEY-1 rather than NOTCH. Furthermore, the MAP-kinase pathway has a negative effect on both γ-secretase expression and activity. Only the exogenous expression of NOTCHIC had a stimulatory effect on tumor growth, but it is still unable to promote a progression from PTC to ATC. NOTCH is not required for tumor formation as RBP-Jκ deletion failed to produce a phenotype. The findings suggest a potential role of NOTCH as a modulator, which could, in part, explain the contradictory previous data on the role of NOTCH in thyroid cancer.
Footnotes
Acknowledgments
Special thanks to Prof. Martin McMahon (Huntsman Cancer Center, Utah) for the BrafCA mice, Prof. Freddy Radtke (EPFL, Lausanne) for the RosaNotch mice, and Prof. Tasuku Honjo (University of Kyoto, Japan) for the Rbp-jκKO mice. A special thank you to Laurent Lecomte and Momirka Trenkoska-Olmo for the monitoring of the mice. Also, we would like to acknowledge Alexandre Sarre at the Cardiovascular Assessment Facility of the University of Lausanne (CAF) for the ultrasound images. Furthermore, we would like to acknowledge the Microscopy Imaging Center of the University of Bern (MIC). Florian Traversi is enrolled in the Graduate School for Cellular and Biomedical Sciences (GCB) of the University of Bern.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Swiss National Foundation grant 31003A_149824/1. The laboratory of RPC is also supported by the Swiss National Science Foundation grant NCCR-TransCure.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2