
Abstract
Background:
Tachycardia, cardiac hypertrophy, and elevated body temperature are major signs of systemic hyperthyroidism, which are considered to reflect the excessive thyroid hormone (TH) action in the respective peripheral tissues. However, recent observations indicate that the central actions of TH also contribute substantially to cardiovascular regulation and thermogenesis.
Methods:
In this study, we dissect the individual contributions of peripheral TH action versus the central effects in body temperature regulation and cardiovascular functions by taking advantage of mice lacking the TH transporters monocarboxylate transporter 8 (MCT8) and organic anion transporting polypeptide 1C1 (OATP1C1) (M/O double knock-out [dko]), which exhibit elevated serum triiodothyronine (T3) levels while their brain is in a profoundly hypothyroid state. We compared these animals with wild-type (WT) mice that were treated orally with T3 to achieve similarly elevated serum T3 levels, but are centrally hyperthyroid. For the studies, we used radiotelemetry, infrared thermography, gene expression profiling, Western blot analyses, and enzyme linked immunosorbent assays (ELISA) assays.
Results:
Our analyses revealed mild hyperthermia and cardiac hypertrophy in T3-treated WT mice but not in M/O dko animals, suggesting that central actions of TH are required for these hyperthyroid phenotypes. Although the average heart rate was unaffected in either model, the M/O dko exhibited an altered heart rate frequency distribution with tachycardic bursts in active periods and bradycardic episodes during resting time, demonstrating that the stabilization of heart rate by the autonomic nervous system can be impaired in centrally hypothyroid animals.
Conclusions:
Our studies unravel distinct phenotypical traits of hyperthyroidism that depend on an intact central nervous system, and provide valuable insight into the cardiovascular pathology of the Allan–Herndon–Dudley syndrome, a condition caused by the lack of MCT8 in humans.
Introduction
Thyroid hormones (THs), the prohormone thyroxine (3,3′,5,5′-tetraiodothyronine; T4) as well as the receptor-active form triiodothyronine (3,3′,5-triiodothyronine; T3), are essential for development and maintenance of almost all tissues. Consequently, TH availability is tightly controlled, as dysregulation can lead to a wide variety of symptoms. In hyperthyroidism, patients often show tachycardia and cardiac hypertrophy (1), weight loss (2,3), and heat intolerance (4), while opposite effects are observed in hypothyroidism, including bradycardia (1), weight gain (2,5), and cold intolerance (4).
According to the prevailing view, these symptoms reflect the elevated or reduced direct actions of TH in the respective peripheral tissues. Recently, however, this concept has been challenged by studies suggesting an important contribution of central TH actions to the phenotypes observed in hyperthyroidism (6 –10): activity of the interscapular brown adipose tissue (iBAT), which is the main site for nonshivering thermogenesis (11,12), was found to be induced by T3 through the ventromedial hypothalamus that in turn activates the sympathetic nervous system (SNS) and triggers facultative thermogenesis (6,8). Likewise, TH actions in the anterior hypothalamic area were mandatory for the correct establishment of the autonomic control of cardiovascular functions (10,13), and TH signaling was also required in the paraventricular nucleus of the hypothalamus to induce hepatic endogenous glucose production (14). Collectively, these findings suggested that central TH action might represent a crucial component for proper peripheral functions. However, to dissect peripheral versus central TH action, animal models are needed that allow to modulate the TH status in brain versus peripheral organs separately.
In this respect, the establishment of mouse models that are deficient in specific TH transporters and, therefore, showing an altered tissue-specific TH state has opened up new experimental approaches. Mice deficient in the monocarboxylate transporter 8 (MCT8) exhibit elevated serum T3 concentrations, causing a hyperthyroid condition in liver, kidney, white adipose tissue (WAT), and skeletal muscle, while their central nervous system (CNS) shows a mild hypothyroid state due to decreased serum T4 concentrations and an impaired transport of T3 into the brain (15 –19).
MCT8-deficient mice lacking additionally the T4-specific transporter organic anion transporting polypeptide 1C1 (OATP1C1, so-called M/O dko mice) display an even more profound TH deprivation in the CNS, since in the absence of both TH transporters the passage of T4 and T3 across the blood–brain barrier is strongly diminished (20). As a consequence, M/O dko mice exhibit a disturbed neuronal development and locomotor impairments, reflecting the phenotype of Allan–Herndon–Dudley syndrome (AHDS) patients, which carry inactivating mutations in MCT8 (21). In peripheral tissues, however, M/O dko mice display the same hyperthyroid phenotype as Mct8 knock-out (ko) mice, as Oatp1c1 expression is largely restricted to the CNS (22,23).
In this study, we took advantage of the unique imbalance between peripheral and central TH action in M/O dko mice to dissect central versus peripheral action of TH on the cardiovascular system and thermoregulation. By comparing the animals with wild-type (WT) mice that were treated with T3 to achieve similarly elevated serum T3 levels as Mct8 ko and M/O dko animals, we were able to dissect the individual contribution of brain and periphery for different phenotypical aspects of hyperthyroidism, as the brain of T3-treated WT animals is in a hyperthyroid state, while the M/O dko brain has been hypothyroid for their entire postnatal life. We found that hyperthermia as well as cardiac hypertrophy as characteristic features of hyperthyroidism do require a central contribution. Moreover, while average heart rate was not significantly affected in M/O dko mice, their heart rate frequency distribution was much broader than in the T3-treated WT animals, indicating a defect in the central autonomic control.
Materials and Methods
Animal husbandry and study design
Oatp1c1fl/fl (served as WT control), Oatp1c1 ko (24), Mct8 ko (15), and Mct8/Oatp1c1 dko (20) mice have been previously described, and for our study adult males on a C57Bl/6 background were used. WT C57Bl/6NCr mice at the age of 4–5 months were purchased from Charles River and rendered hyperthyroid by supplying T3 (T6397; Sigma-Aldrich, Germany) for 14 days in drinking water containing 0.01% bovine serum albumin (BSA) (A7906; Sigma-Aldrich)—a treatment duration sufficient to result in stable serum concentrations (25). The individual dose of T3 was adjusted according to the water intake of the animal to 800 ng/day, resulting in T3 serum levels comparable with M/O dko mice (18,26). Animals were single housed on a 12-hour light/dark cycle with an ambient temperature of 22°C ± 1°C with nesting material and ad libitum access to food (#1324; Altromin) and water. All animal procedures were approved by the Ministerium für Energiewende, Landwirtschaft, Umwelt, Natur und Digitalisierung, Schleswig-Holstein, Germany.
Infrared thermography
Infrared thermography pictures were taken with a Infrared-Thermal-Imaging-Camera (FLIR systems T355; Sweden) from the tail and iBAT area in freely moving animals. For 32°C and 38°C, animals were placed onto a heated platform for 10 minutes. For cold exposure, animals were kept at 10°C for 16 hours before infrared thermography.
Radiotelemetry and blood pressure analysis
For recording heart rate, body temperature, and activity in freely moving mice an implantable telemetry system with transmitters and receivers (G2-HR E-Mitter and ER-4000; Respironics) was used (9,10). Mice were anesthetized using isoflurane and the transmitter was implanted into the abdominal cavity with the electrodes sutured to the lower left chest and right shoulder. For pain relief, mice were intraperitoneally (i.p.) injected for 2 days with 5 mg/kg body weight Rimadyl [carprofen, 2-(6-chloro-9H-carbazol-2-yl)propanoic acid; Pfizer, Berlin, Germany]. After 7 days of recovery, baseline was recorded for 72 hours. The recordings were averaged for 30 minutes and subsequently the mean for all 3 days was calculated. Next, the mice were injected i.p. with saline (5 μL/g body weight; Berlin-Chemie AG, Germany) and the highest absolute heart rate (bpm) achieved within 20 minutes after injection was taken. To quantify autonomic input, the mice were injected with scopolamine methyl bromide (0.1 mg/kg body weight i.p.; Sigma-Aldrich) to block parasympathetic nervous system (PSNS) activity and 40 minutes later with timolol maleate (1 mg/kg body weight i.p.; Sigma-Aldrich) to estimate the SNS activity. For the animals with induced hyperthyroidism, baseline was recorded from day 9 to day 12 of T3 treatment with injections on day 12 and 13. For blood pressure measurement, a noninvasive tail-cuff system was used (Model SC-100; Hatteras Instrument, Cory, NC).
Gene expression analysis
RNA isolation was performed using RNeasy Lipid Tissue Kit (QIAGEN, Germany) for iBAT, and inguinal WAT (iWAT), and the RNeasy Mini Kit (QIAGEN) for liver, brain, and heart. cDNA synthesis was performed using the RevertAid cDNA Kit (Thermo Fisher Scientific, Germany) with oligo (dT)18 primers. Quantitative polymerase chain reaction (qPCR) analysis was performed using SYBR green master mix (+Rox, Roche, Germany) and Quantstudio Applied Biosystems (Thermo Fisher Scientific). For each tissue, three housekeeping genes were tested and the most stable combination of reference genes was determined using Normfinder (27), namely hypoxanthine-guanine phosphoribosyltransferase (Hprt) for iBAT, Cyclophilin and ribosomal protein, large, P0 (Rplp0) for iWAT, ribosomal protein L32 (Rpl32) for heart, and Hprt and Cyclophilin for brain or liver. Results were calculated using the ΔΔCT method, and standard curves were recorded to correct for PCR efficiency. Primer sequences are provided in Supplementary Table S1.
Western blot
Proteins of iBAT homogenates were separated on SDS Gels (TGX Stain-Free Fast Cast 12%; Bio-Rad Laboratories, Germany) and transferred onto a polyvinylidene fluoride membrane (IPVH00010 size 0.34 μm; Merck Millipore, Cork, Ireland). For the detection, primary antibodies [anti-UCP1; 1:10,000 (28), anti-OXPHOS cocktail (1:2000, 45-8099; Thermo Scientific), anti-β-actin (1:10,000, A1978; Sigma-Aldrich)] and secondary antibodies (anti-rabbit or anti-mouse polyclonal HRP-conjugated antibodies, P0448 or P0447; DAKO, Denmark) were used. Chemiluminescence was recorded using ChemiDoc (Touch Imaging System; Bio-Rad Laboratories) and quantified using ImageLab Software (Bio-Rad Laboratories). Protein expression was normalized to β-actin.
ELISA
Serum levels of total T4 (EIA-1781; DRG Instruments GmbH, Germany) and total T3 (DNOV053; NovaTec Immundiagnostica GmbH, Germany) were measured using commercial available ELISA kits. Furthermore, cyclic adenosine monophosphate (cAMP) levels in iBAT were determined according to manufacturer's instruction (RPN225; GE Healthcare, United Kingdom).
Contractile response studies using myograph
A wire-myograph (520A-DMT; AD Instruments, Oxford, United Kingdom) was used to measure contractility of the aorta as described previously (29,30). Increasing concentrations of phenylephrine (P6126; Sigma-Aldrich; 10−8–10−2 M) were added in three-minute intervals to perform a dose–response curve. The force was detected and normalized by the previously recorded KCl stimulation force using Labchart software 8.1 (ADInstruments, Oxford, United Kingdom).
Ex vivo lipolysis
Pieces of iBAT (2–5 mg), iWAT (10–15 mg), and gonadal WAT (gWAT) (10–15 mg) were incubated for 2 hours in lipolysis buffer (REF-21063-029; Life Technologies, Darmstadt, Germany) with 2% BSA (A7906; Sigma-Aldrich) at 37°C and 5% CO2. Afterward, the supernatant was diluted in free glycerol reagent (F6428; Sigma-Aldrich) and measured at 540 nm using SPECTROstar Nano Microplate Reader (BMG Labtech, Germany) with a standard control (G7793; Sigma-Aldrich).
Hepatic glycogen content determination
Glycogen content determination on dry-ice frozen liver tissue samples was performed as described previously (31).
Thermal hot bridge analysis
Skin samples still containing fur were dissected from the interscapular area of WT, Mct8 ko, Oatp1c1 ko, and M/O dko mice, and analyzed using a transient hot bridge instrument (Linseis, Selb, Germany) to calculate heat conductivity, diffusivity, and capacity.
Statistical analysis
For data analysis Microsoft Office Excel and GraphPad Prism 5 and 7 (San Diego, CA) were used. Values are shown as mean ± standard error of the mean. Statistical testing was performed using two-way analysis of variance (ANOVA) with Bonferroni posthoc test (for studies with four genotypes or time profiles), and paired or unpaired Student's t-test with Welch's correction (for T3-treatment studies). Heart rate frequency distribution and body temperature in the T3-induced model were analyzed with two-way ANOVA with Holm–Sidak's multiple comparisons test. p-Values <0.05 were considered significant (Supplementary Table S2).
Results
To examine the impact of a TH-deficient CNS on the peripheral symptoms of hyperthyroidism, we performed a comprehensive thermoregulatory and cardiovascular analysis of M/O dko mice in comparison with WT and the respective Mct8 ko and Oatp1c1 ko single mutant mice (Fig. 1A). In line with previous reports (15,20), serum TH values were normal in Oatp1c1 ko, whereas Mct8 ko and M/O dko mice exhibited the expected changes with elevated serum T3 and low serum T4 (Fig. 1B). To identify effects caused by the elevated T3 serum levels, we simultaneously studied WT animals that received 800 ng/day T3 through the drinking water for 14 consecutive days in a second experiment (Fig. 1A), resulting in a similarly altered serum TH profile (Fig. 1B). This dose was sufficient to reach the brain and induce the expected changes in gene expression of hairless (Hr), deiodinase type 3 (Dio3) and kruppel-like factor 9 (Klf9) (Supplementary Fig. S1A). Body weight and food intake were assessed in all experimental groups (Fig. 1C–F) and confirmed significantly reduced body weight in M/O dko mice, while Oatp1c1 ko, Mct8 ko as well as T3-treated WT mice did not differ significantly from the control groups (Fig. 1C, D and Supplementary Table S2). Relative food and water intake were increased in M/O dko and T3-treated WT mice (Fig. 1E, F and Supplementary Fig. S1B), whereas locomotor activity was comparable in all experimental groups (Fig. 1G, H and Supplementary Table S2).
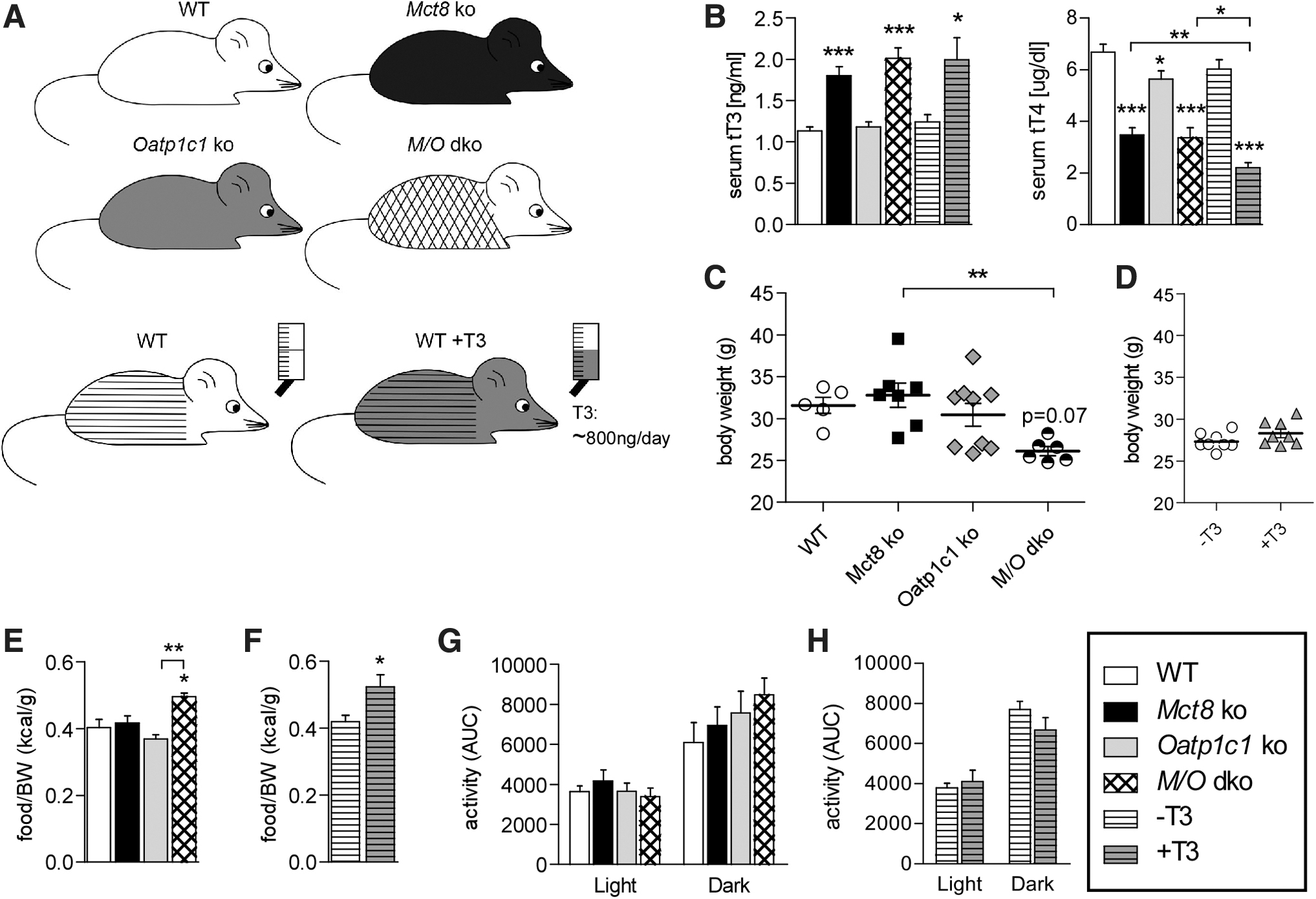
(
For monitoring body temperature, 72 consecutive hours were recorded by radio telemetry in freely moving and conscious animals, averaged and plotted in a 24-hour profile. The data did not reveal any differences in the TH transporter-deficient mice (Fig. 2A, B and Supplementary Table S2); however, T3-treated WT mice exhibited a mildly but significantly increased body temperature during the light (inactive) but not the dark (active) phase (Fig. 2C, D, p Interaction = 0.0013, two-way ANOVA). Subsequently, infrared pictures were taken from different body areas to assess heat production and heat loss, but showed no significant difference in iBAT (Fig. 2E, F and Supplementary Table S2) or tail temperatures (Fig. 2G, H and Supplementary Table S2). Interestingly, when the mice were cold challenged at 10°C for 16 hours, mice lacking Oatp1c1 displayed a minor reduction in body but not iBAT or tail temperature (Supplementary Fig. S1C–E and Supplementary Table S2), suggesting a mild cold sensitivity. Moreover, we observed an elevated tail temperature in Mct8 ko mice at 32°C (Supplementary Fig. S1E and Supplementary Table S2), indicative of mild heat stress. Analysis of heat conductivity, diffusivity, and capacity of the skin using transient hot bridge did not disclose any differences between the groups (Supplementary Table S3), suggesting normal skin and fur insulation properties. Taken together, these data suggest no major impairments in body temperature regulation at the phenotypical level caused by the lack of Oatp1c1 or Mct8; however, despite their elevated serum T3, M/O dko mice did not display the mild daytime hyperthermia observed in T3-treated WT mice.
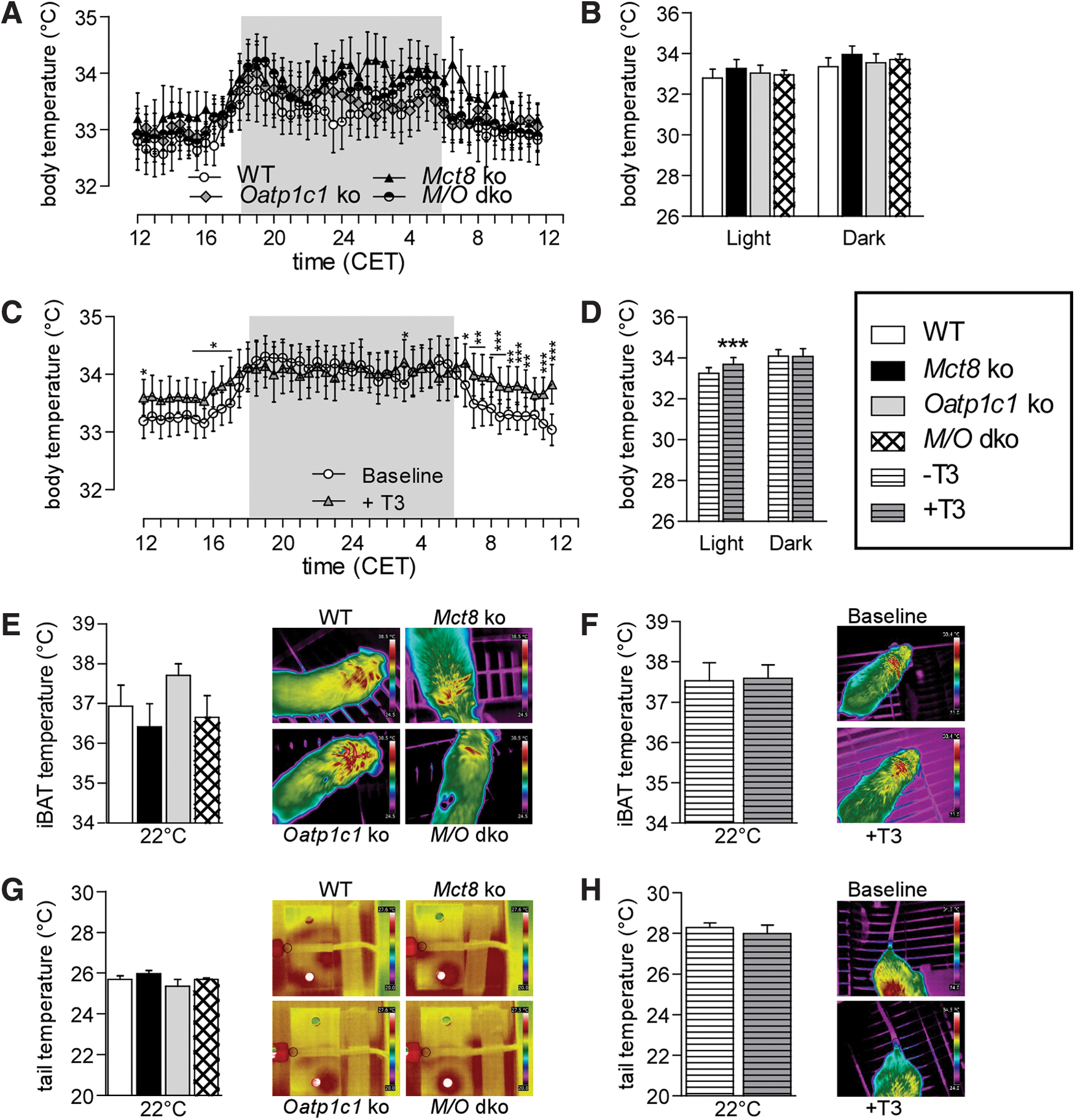
(
To identify alterations in body composition, distinct fat depots were analyzed (Fig. 3A, B). In TH transporter-deficient mice, no alterations in iBAT weight were detected, whereas T3-treated WT mice exhibited a significant increase in iBAT weight compared with controls (Fig. 3B), consistent with previous studies (25). Normalized iWAT and gWAT weights were not different in T3-treated WT, but gWAT was lighter in M/O dko mice compared with Oatp1c1 ko animals (Fig. 3A, B)
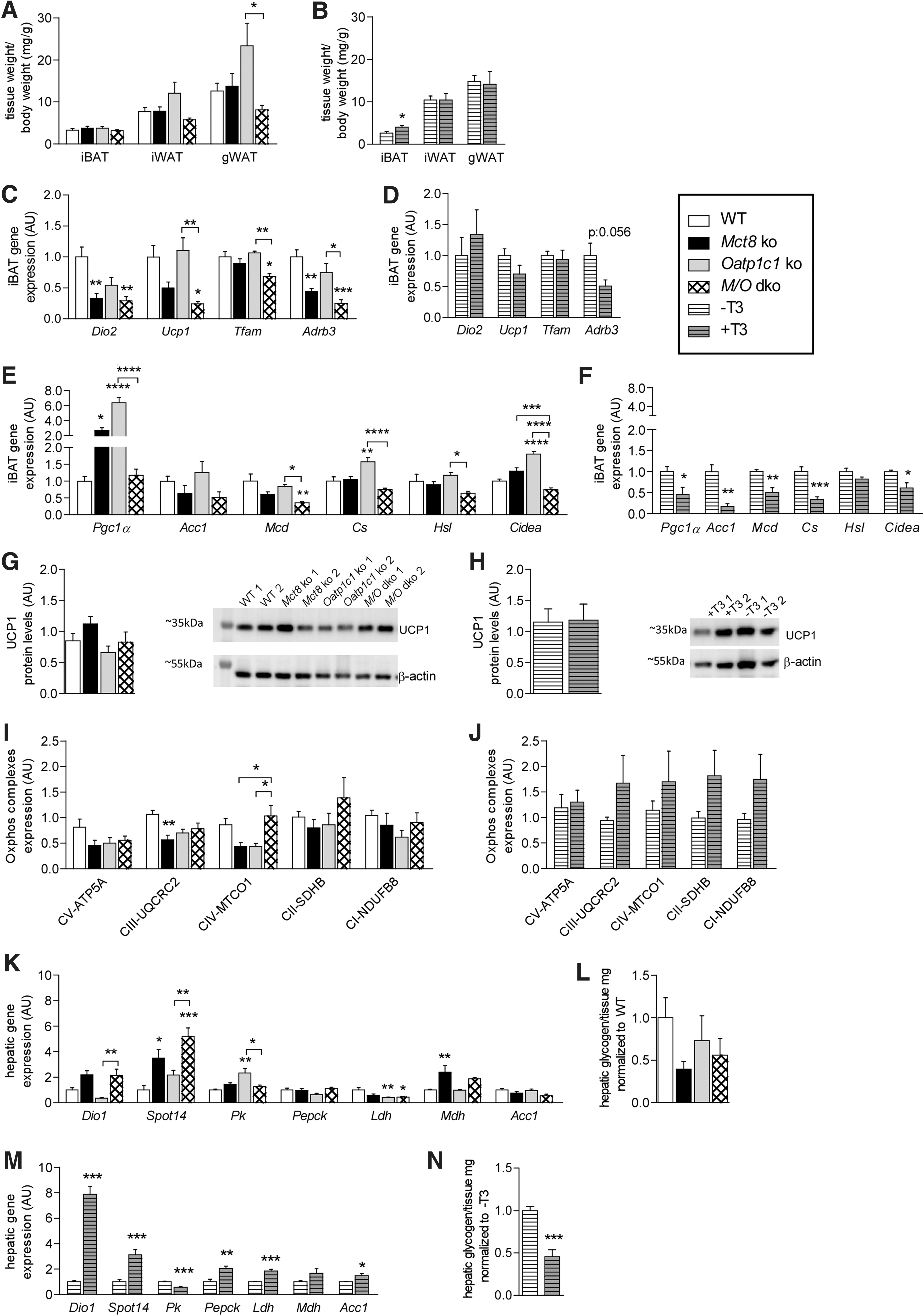
(
To identify molecular alterations in thermogenesis, gene expression analysis in iBAT was performed, revealing that uncoupling protein 1 (Ucp1), iodothyronine deiodinase 2 (Dio2), mitochondrial transcription factor A (Tfam), and adrenergic receptor beta 3 (Adrb3) were significantly reduced in M/O dko mice, while Dio2 and Adrb3 mRNA were downregulated in Mct8 ko mice (Fig. 3C and Supplementary Table S2). In T3-treated WT mice, only a trend toward reduced Adrb3 transcript levels was observed (Fig. 3D). However, despite the changes in Adrb3 mRNA, the intracellular iBAT cAMP levels were comparable between all groups, indicative of similar sympathetic input (Supplementary Fig. S1F, G). Expression analysis of genes involved in fatty acid metabolism in iBAT revealed an overall reduction in the M/O dko mice, whereas Oatp1c1 ko mice showed an increase in citrate synthase and Cidea (Fig. 3E and Supplementary Table S2). In T3-treated iBAT, a reduced expression of most fatty acid metabolic genes was noted (Fig. 3F). Despite the respective changes in mRNA expression, UCP1 protein levels were not significantly different in any group (Fig. 3G, H and Supplementary Table S2). Analysis of protein levels of selected oxidative phosphorylation complex proteins revealed a reduction in complex III in Mct8 ko mice, while complex IV was increased in M/O dko mice as compared with Mct8 or Oatp1c1 single ko mice (Fig. 3I and Supplementary Table S2). There was no effect of T3 treatment (Fig. 3J). Overall, the analysis of iBAT showed a minor reduction in the thermogenic profile of Mct8 ko mice, which was more pronounced in the M/O dko mice, while the iBAT of T3-treated WT mice was largely normal.
In iWAT, another adipose depot potentially capable of thermogenesis, we detected increased Dio2 transcript levels in the Mct8 ko mice but no consistent changes in Ucp1, Cidea, peroxisome proliferator-activated receptor gamma coactivator 1 alpha (Pgc1α) and malonyl-CoA decarboxylase (Mcd) mRNA expression (Supplementary Fig. 1H and Supplementary Table S2). Likewise, T3-treated WT mice also did not show any changes in Ucp1 mRNA, despite a fivefold elevation of Dio2 mRNA (Supplementary Fig. S1I), suggesting that the T3 levels are not sufficiently high to induce significant browning. No changes were observed for genes involved in iWAT fatty acid metabolism (Supplementary Fig. S1J and Supplementary Table S2) and only a minor reduction in lipoprotein lipase (Lpl) mRNA in the T3-treated WT was observed (Supplementary Fig. S1K). On the functional level, lipolytic activity was only mildly elevated in iBAT and iWAT but not in gWAT of the M/O dko mice (Supplementary Fig. S1L and Supplementary Table S2). Taken together, these data show only minor alterations in fatty acid metabolism.
We then analyzed liver metabolism, which is well known to be regulated by direct (32) and central T3 actions (14). Analysis by qPCR confirmed the expected increase in the established T3 target genes Dio1 and TH responsive (Spot14) in Mct8 ko mice (15) as well as the T3-treated animals. Transcript levels of genes involved in glucose metabolism were not consistently altered in Mct8 ko mice (Fig. 3K and Supplementary Table S2), and hepatic glycogen content was similar in all groups (Fig. 3L and Supplementary Table S2). In contrast, T3-treated WT mice had a reduced pyruvate kinase (Pk) expression with increased phosphoenol pyruvate carboxykinase (Pepck) and lactate dehydrogenase (Ldh) mRNA (Fig. 3M), which together with the reduced hepatic glycogen content (Fig. 3N) reflects the increased glucose demand.
As the heart is another important T3 target tissue (33), we performed a detailed cardiovascular analysis. Surprisingly, we found no hypertrophy in Mct8 ko and M/O dko mice (Fig. 4A and Supplementary Table S2), although this would have been expected from elevated serum T3 as seen in the T3-treated WT mice (Fig. 4B). We subsequently analyzed heart rate in vivo using the implantable radio transmitter in freely moving and conscious animals. No differences were observed in the 24-hour profile and the area under the curve in light and dark phase, neither in the transporter ko mice nor in the T3-treated animals (Fig. 4C, D; Supplementary Fig. S2A, B and Supplementary Table S2), suggesting that the T3 serum levels are not sufficient to induce tachycardia. However, the basal heart rate distribution during the dark and light phase revealed significant changed frequency percentages exclusively in M/O dko mice, characterized by an overall broader distribution with increased lower and higher frequencies, while the medium heart rate frequencies were reduced (Fig. 4E and Supplementary Fig. S2C). This was not observed in the T3-treated mice (Fig. 4F and Supplementary Fig. S2D). Most remarkably, the tachycardic episodes in the M/O dko mice correlated with periods of locomotor activity, while bradycardia occurred at rest (Fig. 4G)—an effect much less pronounced in the T3-treated animals (Fig. 4H). The stress response to a single saline injection, however, did not differ between the genotypes or in T3-treated WT (Fig. 4I, J and Supplementary Table S2), and likewise, the acute pharmacological denervation by injecting scopolamine and timolol revealed no significant differences in the sympathetic and parasympathetic input to the heart (Fig. 4K, L and Supplementary Table S2). The intrinsic heart rate after full pharmacological denervation was also not different between the different genotypes (Fig. 4M and Supplementary Table S2), but was higher in the T3-treated WT compared with controls (Fig. 4N). In concordance, genes involved in the regulation of cardiac properties and regulated by TH were not significantly altered between the genotypes, and only phospholamban (Pln) and brain natriuretic factor (Bnf) were reduced in the T3-treated WT mice (Supplementary Fig. S2E, F). However, there were no significant differences in the ratios between α and β-Mhc mRNA levels in either groups (Supplementary Fig. S2F). Blood pressure as recorded by tail cuff and aortic contractility as measured by wire myography displayed no significant differences between the genotypes (Supplementary Fig. S2G, H). Taken together, these data show no obvious endogenous cardiac defects caused by the lack of OATP1C1 or MCT8, presumably due to the compensation by the highly expressed MCT10 (Supplementary Fig. S2I); however, although the central stress response as well as the SNS and PSNS activity seem to work normally on average, the altered heart rate frequency distribution clearly demonstrates a less stable central control and bursts of tachycardic episodes correlating with high activity.
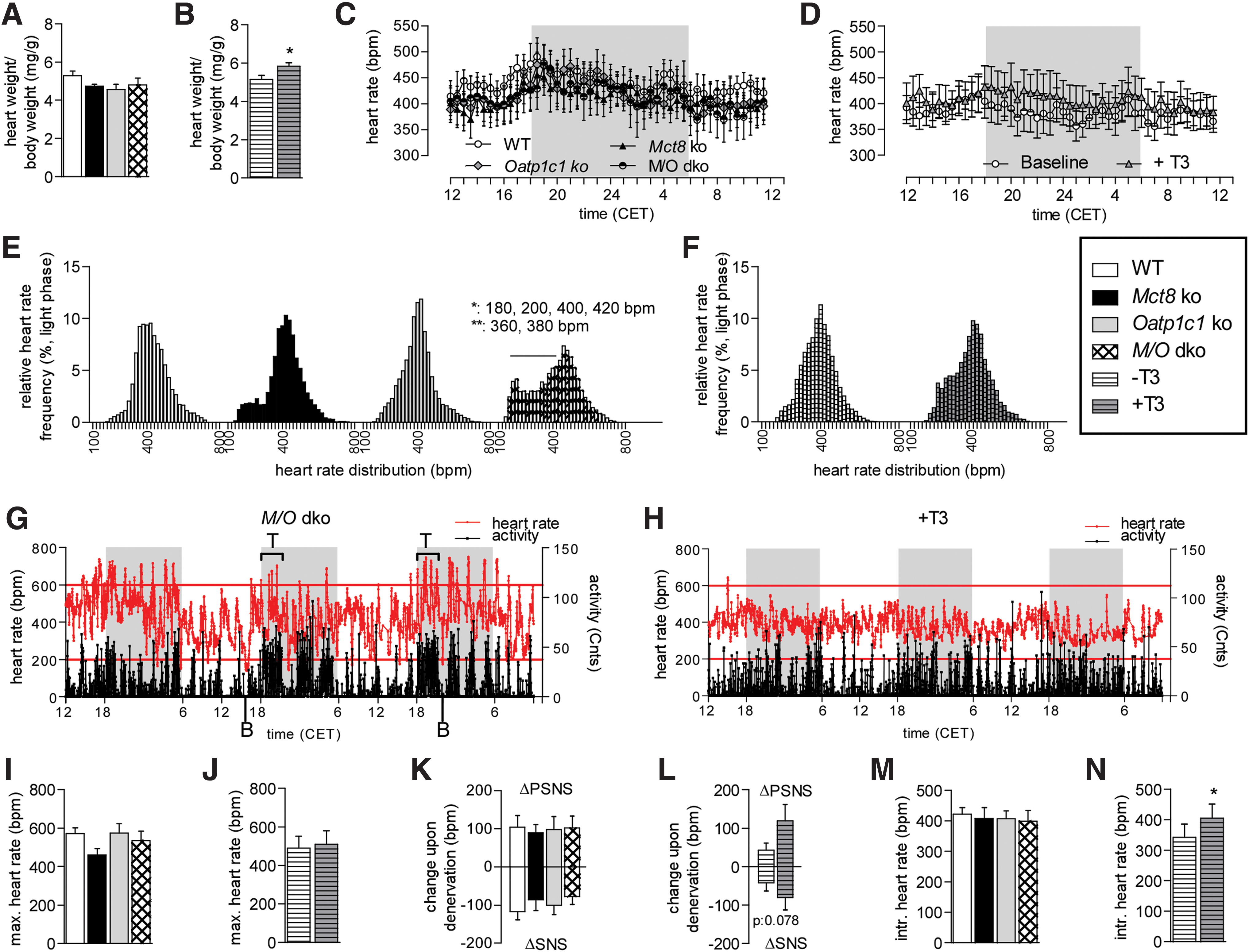
(
Discussion
In our comprehensive cardiovascular and thermoregulatory phenotyping of M/O dko mice, we observed several abnormalities that were not found in WT mice with a similar T3/T4 serum profile. In principle, there are three possibilities that can account for these differences: (i) the lack of transporters in peripheral tissues impairs the TH import resulting in a hypothyroid state, (ii) the hypothyroid brain of M/O dko animals causes a difference in the central control, which contrasts with the hyperthyroid brain of T3-treated WT mice, or (iii) a developmental defect in the brain of M/O dko mice leads to a permanent alteration in the central control.
Does the lack of Mct8 and/or Oatp1c1 limit TH uptake in the peripheral tissues? Given the inconspicuous phenotype of Oatp1c1 ko mice, which only had problems with maintaining their body temperature upon cold exposure [similar to what is observed in the first patient with a loss of OATP1C1 (34)], and the restricted expression pattern of Oatp1c1 largely in the brain (22,23), it seems unlikely that the loss of this transporter causes any TH uptake defects in peripheral tissues. Likewise, the lack of MCT8 did not result in any hypothyroid-like conditions in the investigated peripheral tissues, as shown by elevated Dio1 in liver and reduced Dio2 in iBAT (15,35). Similarly in the heart, the T3 sensitive pacemaker channels Hcn2 or Hcn4 (36) were not reduced, but even mildly elevated, and no bradycardia was observed, which would be expected in hypothyroidism. These findings concur with previous studies in Mct8 ko mice, showing higher T3 levels in liver, kidney, and skeletal muscle, and normal T3 levels in iBAT (15,18). Taken together, this suggests that the M/O dko phenotype is likely not caused by an impaired TH uptake in liver, fat, or heart—presumably due to the compensation by other TH transporters such as MCT10 (37).
THs can regulate the activity of liver, heart, or iBAT by acting not only directly in the target tissue, but also indirectly through modulation of the autonomic nervous system. Recent studies have shown that central T3 injections often replicate the phenotype observed in systemic hyperthyroidism, including elevated endogenous hepatic glucose production (14) or increased iBAT thermogenesis (6,8), suggesting that a hyperthyroid brain synergistically enhances the peripheral actions of the hormone. By comparing the phenotype of the M/O dko mice with WT animals with similarly elevated serum T3 levels, we obtained valuable insight into the consequences of a hypothyroid brain for the function of liver, iBAT, and heart: Any defect that is observed in M/O dko mice but not T3-treated WT is likely caused by the impaired TH signaling in the brain.
However, without an inducible M/O dko animal model it is not possible to distinguish whether a defect such as the altered heart rate frequency distribution results from acutely impaired TH action in the brain or from a defect arising from hypothyroidism during development. Nevertheless, the animal model reflects the combination of acute and developmental hypothyroidism also found in the AHDS patients. Moreover, any phenotype found in T3-treated adult WT but not M/O dko mice is likely a consequence of acute TH actions in the brain, as both groups have similarly elevated peripheral T3 but strongly differ in their uptake of TH into the brain.
What are the central contributions? With regard to the liver, we observed a switch from glycolysis to gluconeogenesis together with reduced hepatic glycogen stores in the T3-treated animals as expected from previous studies (25,38), suggesting increased hepatic glucose production. However, these changes were not observed in Mct8 ko or M/O dko mice, indicating that the brain is required for the induction of hepatic glucose production, as previously observed (14).
TH also influences temperature homeostasis as seen by the heat intolerance in hyperthyroid individuals (39). The classic concept proposed direct effects of TH on obligatory thermogenesis in muscle (40) and on facultative thermogenesis in iBAT (39); however, recent rodent studies challenged this paradigm by showing that central TH action might be equally important for iBAT activation (6,7). Accordingly, we observed a mildly elevated body temperature in the daytime resting period in our T3-treated WT mice as expected (6,25), while this was not found in M/O dko mice. Instead, we noticed a lower expression of thermogenic markers such as Ucp1 or Tfam in the M/O dko iBAT and a tendency toward reduced iBAT thermogenesis in the infrared images, which was not observed in the T3-treated WT, suggesting that M/O dko compensate the T3-induced hyperthermia by a minor reduction in facultative iBAT thermogenesis.
The fact that a similar compensation has not been observed in the T3-treated mice is likely explained by the fact that in these animals the intact central T3 action increases the body temperature set point (25)—an effect that does not occur in M/O dko mice due to their hypothyroid brain and, therefore, they defend a normal body temperature.
A classic hallmark of hyperthyroidism in patients is cardiac hypertrophy, which we also observed in our T3 model—to an extent comparable with classic hypertrophy animal models (41). Surprisingly, M/O dko mice did not develop cardiac hypertrophy despite their elevated serum T3 levels. As cardiac T3 import seems not to be impaired by the lack of MCT8, presumably due to compensation by MCT10, it can be concluded that the T3-induced hypertrophy requires a central contribution. This is supported by previous studies, which revealed that hyperthyroidism did not cause hypertrophy in a transplanted heart (42,43). Likewise, studies using the β-blocker propranolol (44) or mice lacking all three β-adrenergic receptors (45) also suggested that SNS input is crucial for a T3-induced cardiac hypertrophy. Similarly, selective overexpression of human DIO2 in the myocardium, resulting in local hyperthyroidism, lead to tachycardia but not to hypertrophy (46).
Unexpectedly, we did not observe a chronic tachycardia in the Mct8 ko or M/O dko mice, although this is another hallmark of hyperthyroidism (47). However, our T3-treated WT did not show tachycardia either, suggesting that the serum T3 elevation may be insufficient to increase heart rate. This seems plausible as the classical landmark studies in the field employed approximately fourfold higher T3 doses to induce an increase of 25% in heart rate (48). Most remarkably, however, we observed significant changes in the heart rate frequency distribution in the M/O dko mice, including tachycardic bursts that correlated with locomotor activity and bradycardic episodes in resting periods. Because this was not observed in Mct8 ko or T3-treated WT, our results demonstrate that the severely hypothyroid brain of the M/O dko mice is responsible for this phenotype.
These findings are of immediate clinical relevance for the AHDS, as sudden death due to cardiac arrhythmias that often arise from tachycardic bursts is a common cause of death in patients with MCT8 impairments (49). Based on our data, the reason for these tachycardic episodes is likely an impaired central autonomic control triggered by physical activity rather than a primary cardiac defect, which advocates strategies such as Triac treatment to improve central TH action (49) and reduce the risk of arrhythmias. Likewise, the impaired hepatic gluconeogenesis in M/O dko mice as compared with T3-treated WT might be clinically relevant for conditions, in which AHDS patients are not properly fed, such as due to time constraints in hospitals. Finally, it is expected that the T3-induced hyperthermia may be observed in AHDS patients when central TH signaling is restored, but peripheral T3 levels have not been sufficiently lowered.
Footnotes
Acknowledgments
We thank the staff of the Gemeinsame Tierhaltung animal facility for technical assistance, and are grateful to Edward Visser for fruitful scientific discussions.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was funded by the Deutsche Forschungsgemeinschaft (Heisenberg Program MI1242/2-2 to J.M.; SPP1629 “Thyroid TransAct” MI1242/6-1 to J.M. and HE3418/8-1 to H.H.) and the Medical Facility of the University of Lübeck (J14-2018) to R.O.; B.H., L.H., S.G., and S.N. are students of the DFG funded GRK1957 “Adipocyte-Brain-Crosstalk.”
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3