
Abstract
Concomitant thyroid cancer in patients with congenital thyroid dyshormonogenesis (TD) is extremely rare and few cases of differentiated thyroid cancer in patients with TD have been reported thus far. In this study, we describe anaplastic thyroid cancer in a 46-year-old woman with TD who had two germline thyroglobulin (TG) gene mutation, c.3790T>C (p, Cys1264Arg) in exon 17 and a novel c.7070T>C (p.Leu2357Pro) in exon 41 of the TG gene. Two affected younger sisters were also found to have the same TG mutation but not anaplastic thyroid cancer. Any thyroid nodular lesions that develop in patients with TD should be investigated carefully.
Introduction
Congenital hypothyroidism is the most common congenital endocrine disorder, with up to 85% of reported cases associated with thyroid dysgenesis (nongoitrous) (1,2). The other cause of congenital hypothyroidism in 10–15% of reported cases is thyroid dyshormonogenesis (TD), which contributes to goitrous changes in most affected patients. TD is caused by genetic defects in thyroid hormone synthesis and metabolism, including iodide trapping, efflux, and organification, as well as recovery of intrathyroidal iodine and supply of thyroglobulin (TG), which provides a matrix for thyroid hormone synthesis (3). Gene mutations identified in patients with TD include defects in sodium-iodide symporter (SLC5A5), pendrin (SLC26A4), thyroid peroxidase, dual oxidase/dual oxidase maturation factor NADPH oxidase complexes, iodotyrosine deiodinase, and TG (3 –7).
The compensatory mechanisms underlying impaired thyroid hormone synthesis, secretion, and recycling contribute to elevated serum thyrotropin (TSH) concentrations, which can result in a goiter, thyroid nodules, and thyroid cancer (8,9). There have been several previous case reports of thyroid cancer in patients with TD (8,10 –12). With the exception of a single patient who exhibited follicular thyroid cancer with focal anaplastic components (10), these case reports represented differentiated thyroid cancer. Hishinuma et al. (8) reported a relatively high incidence of thyroid cancer in patients with TD, especially those with long-standing goiter. Long-term thyroid hormone deficiency, chronic TSH stimulation, and genetic defects could increase the incidence and aggressiveness of thyroid cancer in patients with TD. In this study, we describe a patient with TD caused by a TG gene mutation who developed anaplastic thyroid cancer, along with two siblings who had an identical TG gene mutation.
Patient
A 46-year-old woman was referred to our hospital for thyroid surgery. She had a large thyroid nodule, and fine needle aspiration at a local clinic had revealed a follicular neoplasm. She experienced compressive symptoms three months after fine needle aspiration and decided to undergo thyroid surgery. She had been receiving thyroid hormone replacement since the age of 14 years, and her medical compliance was good. She exhibited normal growth and cognitive function. At the first visit to our hospital, a thyroid function test (Cobas e601; Roche, Basel, Switzerland) was performed. The patient's serum TSH concentration was 0.205 μIU/mL on 100 μg/day of levothyroxine (LT4) replacement, but her serum TG was undetectable. The patient had two younger sisters, both of whom had also received LT4 therapy since childhood. We performed thyroid ultrasonography (US) of the three sisters, all had a diffuse goiter with heterogeneous echogenicity and internal hyperechoic lesions. One sibling (sibling 1) had a history of thyroid lobectomy for a thyroid nodule at 16 years of age and complete thyroidectomy due to a calcified thyroid nodule. Her pathology showed nodular hyperplasia and fibrocalcific nodules. The US findings of our patient revealed a large heterogeneous and hypoechoic nodule with internal calcification (6 cm) in the right lobe, as well as dense calcification in the left lobe (0.9 cm). Staging imaging studies revealed multiple right paratracheal lymph node, lung, and bone metastases (Fig. 1). The patient underwent total thyroidectomy with central lymph node dissection and pathology showed anaplastic thyroid cancer with Riedel's thyroiditis, which was negative for a BRAF V600E mutation. Lenvatinib therapy was started at a dose of 20 mg/day, 2 weeks after surgery. Ten months after initiation of lenvatinib therapy, the patient had disease progression (new liver metastasis and increased size and number of lung metastasis) and passed away 14 months after the diagnosis of anaplastic thyroid cancer.
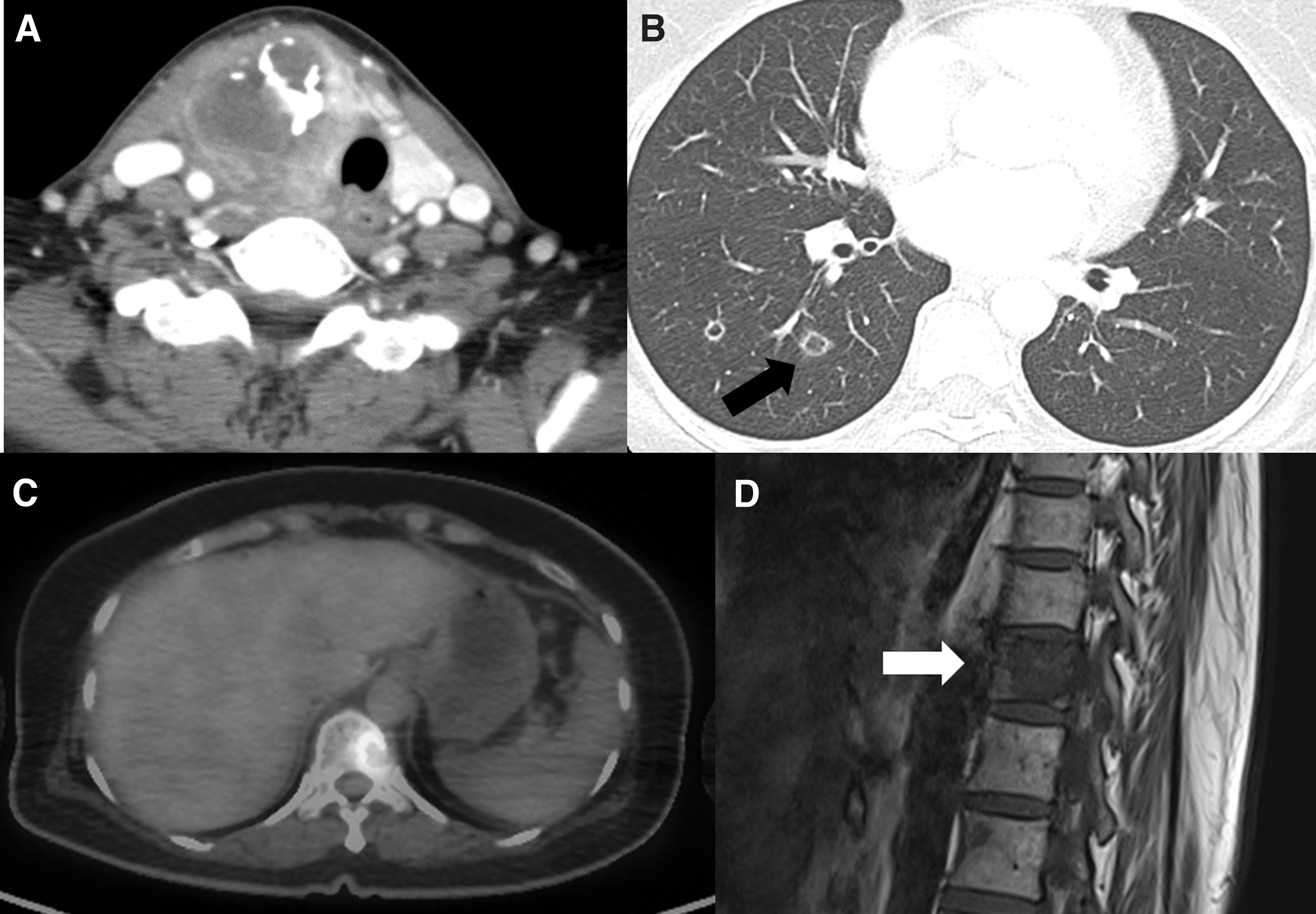
Anaplastic thyroid cancer with multiple lung and bone metastases in the patient. (
Molecular Genetic Studies
This study was approved by the Clinical Review Board of Chonnam National University Hwasun Hospital. Written informed consent was obtained from the patient and both siblings. We evaluated genes associated with thyroid hormone synthesis and transport in the index patient using a targeted gene panel of 23 genes (DUOX2, DUOXA2, FOXE1, GNAS, HESX1, IYD, LHX3, NKX2–1, NKX2–5, PAX8, POU1F1, PROP1, SLC16A2, SLC26A4, SLC5A5, TG, THRA, THRB, TPO, TRH, TRHR, TSHB, TSHR). Exomes were captured using a customized Target Enrichment System Kit (Celemics, Seoul, Korea). Genomic DNA was extracted from peripheral blood sample and targeted gene sequencing was performed with Miseq Platform (Illumina, Inc., San Diego, CA) with 150-bp paired-end sequencing. Sequence reads were aligned to the human reference genome (hg19) using Burrow–Wheeler Alignment MEM algorithm version 0.7.12. SAMtools version 0.1.19, Genome Analysis Toolkits version 3.5, FreeBayes 0.9.2.1, Scalpel-0.5.3, and PICARD tools version 1.96 were used for data analysis. Annotation was performed with a Variant Effect Predictor, dbNSFP.
We found two heterozygous missense mutations in the TG gene in the patient (Fig. 2)—the c.3790T>C mutation has been reported previously in Japanese patients (8) and has a prevalence of 0.02% in the genome aggregation database (gnomAD), while the missense mutation c.7070T>C has not been reported previously. In silico prediction suggested that this novel mutation was deleterious, and that it may be associated with structural and functional changes in the TG protein. Sequencing analysis confirmed identical mutations in both of the patient's affected siblings. Unfortunately, genetic testing of the parents could not be performed as they were not alive, but both of them had no known history of thyroid disease and requiring thyroid hormone replacement. An unaffected brother represented normal thyroid function and US findings, but genetic testing was not performed. The pedigree of this family is shown in Figure 2. There have been 12 previously reported patients with thyroid cancer associated with TD, who had mutations in susceptibility genes (Table 1). To our knowledge, our patient is the first with anaplastic thyroid cancer associated and a TG gene mutation (Table 1).

Family pedigree and DNA sequence analysis of two heterozygous TG gene mutations in the three sisters. (
Summary of Clinical Characteristics of Thyroid Cancer in Thyroid Dyshormonogenesis Patients with Documented Gene Mutations Reported in the Literature, Including Our Case
FTC, follicular thyroid cancer; FVPTC, follicular variant papillary thyroid cancer; NA, not available; PDS, Pendred syndrome; PTC, papillary thyroid cancer; TPO, thyroid peroxidase.
Discussion
TG is a major substrate of thyroid hormone biosynthesis; thus, genetic defects of TG synthesis and secretion could result in congenital hypothyroidism (13). The TG gene has 48 exons and encodes a polypeptide of 2749 residues with 19-amino acid signal peptide (14 –16). Ieiri et al. (17) were the first to demonstrate a TG gene mutation in a patient with congenital goiter; many sporadic and familial TG gene mutations have since been identified (18,19). TG gene mutations show autosomal recessive inheritance, but a higher frequency of familial TG gene mutations have been reported, compared with other gene mutations, in patients with TD (6,8). Structural and functional analyses have shown that some TG gene mutations disrupt the normal three-dimensional structure of TG (14,19,20); other mutations can cause retention of TG in the endoplasmic reticulum (11,21). Our patient and both of her siblings had two heterozygous TG gene mutations. The c.3790T>C missense mutation located at exon 17 involves a cysteine residue of TG; therefore, it is associated with the tertiary structure of the encoded protein product (8,22). The c.7007T>C mutation, in exon 41, has not been reported previously, and further studies are necessary to evaluate its structural and functional implications.
Many case reports of patients with TD indicated the presence of multiple thyroid nodules, large thyroid cysts, and thyroid cancer; however, the mechanisms of thyroid tumorigenesis in these patients remain uncertain (12,23). The predominant hypothesis is that long-term elevated TSH serves as a growth factor for thyroid follicular cells, indirectly affecting tumor activating or suppressive genes (8,11). Previous reports of concomitant thyroid cancer in patients with TD include papillary thyroid cancer and follicular thyroid cancer, with the exception of follicular thyroid cancer with anaplastic components that occurred in a patient with Pendred syndrome (10,11,24,25). Among the 12 previously reported cases of thyroid cancer in TD patients with documented gene mutation, 4 cases (33.3%) had distant metastasis and 6 cases had multifocal disease (10,11,24,25). To the best of our knowledge, this is the first report of anaplastic cancer arising in a patient with TD who had a TG gene mutation.
In conclusion, long-term hypersecretion of TSH in patients with TD may induce the development of thyroid cancer through direct stimulation as a growth factor and indirect changes in thyroid tissue that result in a precancerous fibrotic condition. Thyroid cancer arising from TD is uncommon, but may be highly aggressive. Therefore, careful follow-up of patients with TD is necessary, especially when thyroid nodules are present.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The author(s) did not receive any funding for this work.